Real-Time Time-Dependent Density Functional Theory
Our research group develops innovative methodologies in theoretical chemistry, focusing on tackling the complex problems that drive our work. Recently, we have honed our efforts on creating a unified framework for numerical simulations, specifically designed to model the ionizing irradiation of matter, with a special emphasis on biological systems like DNA and proteins. Through our cutting-edge developments, we make key tools available to the scientific community via the deMon2k program. We welcome collaborations at any level—reach out and join us in pushing the boundaries of scientific discovery!

Theoretical modeling of the physical and physico-chemical stages of ionizing irradiation of matter demands an accurate description of the electron cloud’s response on ultrashort timescales—spanning the attosecond (10⁻¹⁸s) and femtosecond domains. To meet this challenge, we are developing advanced first-principles simulation methods rooted in density functional theory (DFT), striving for exceptional performance to capture the complexity of the molecular systems under study. These systems are often nanometric in scale, with intricate structural and chemical inhomogeneities.
Our approach centers on an innovative Auxiliary DFT framework integrated into the deMon2k program. The RT-TD-ADFT method we are developing is highly versatile, seamlessly interacting with several key functions of deMon2k, including Ehrenfest molecular dynamics (Ehrenfest MD) and hybrid Quantum Mechanics/Molecular Mechanics (QM/MM) approaches. This combination allows us to model complex systems with remarkable accuracy, enabling groundbreaking insights into ionizing radiation’s impact on molecular and biological structures.
The program can be used to simulate the irradiation of matter by fast ions or photons (UV-Vis, XUV, IR…). A recent journal article (2023) summarizes the current program capabilities.
The program can be downloaded from Zenodo (10.5281/zenodo.8301468) for use on CPU or CPU/GPU machines.
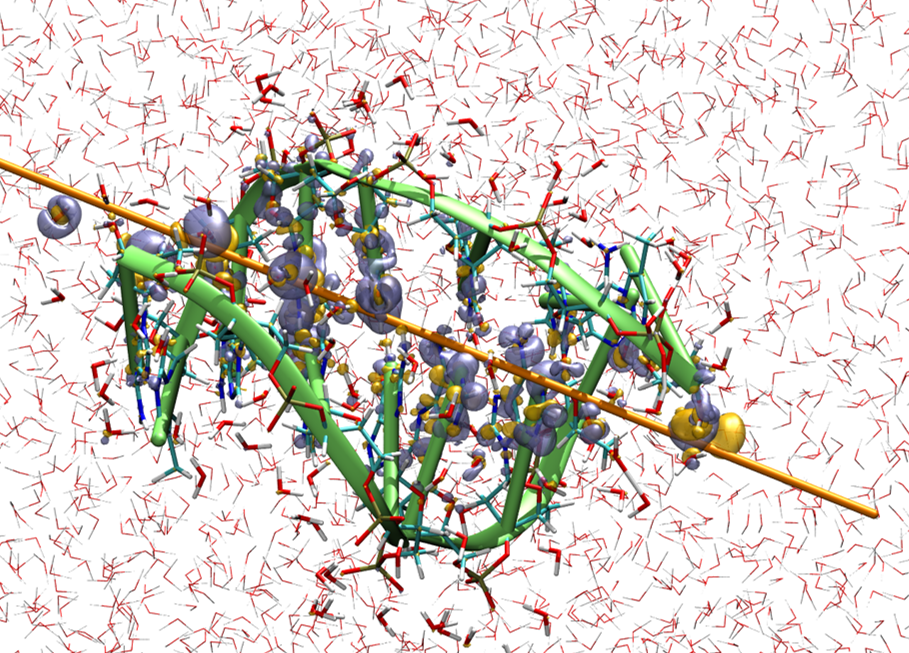
This image, taken from a paper published in 2020, shows the deformation of the electron cloud within a solvated DNA double strand during the passage of a high-energy alpha particle. RT-DT-ADFT simulations have enabled us to understand key elements in the mechanism of energy deposition by ions in DNA.
Multicomponent Dependent Density Functional Theory
Multicomponent DFT (MDFT) is an extension of electronic DFT to molecular systems comprising more than one type of quantum particle. In principle, this innovative approach makes it possible to account for nuclear quantum effects or molecules in intraction with “exotic” particles (muons, antiprotons, positrons…). We have developed a method of this type within the framework of Auxiliary DFT in the deMon2k software. This work is now continuing in collaboration with Prof. Lars Pettersson and Dr. Felix Moncada (Stockholm).
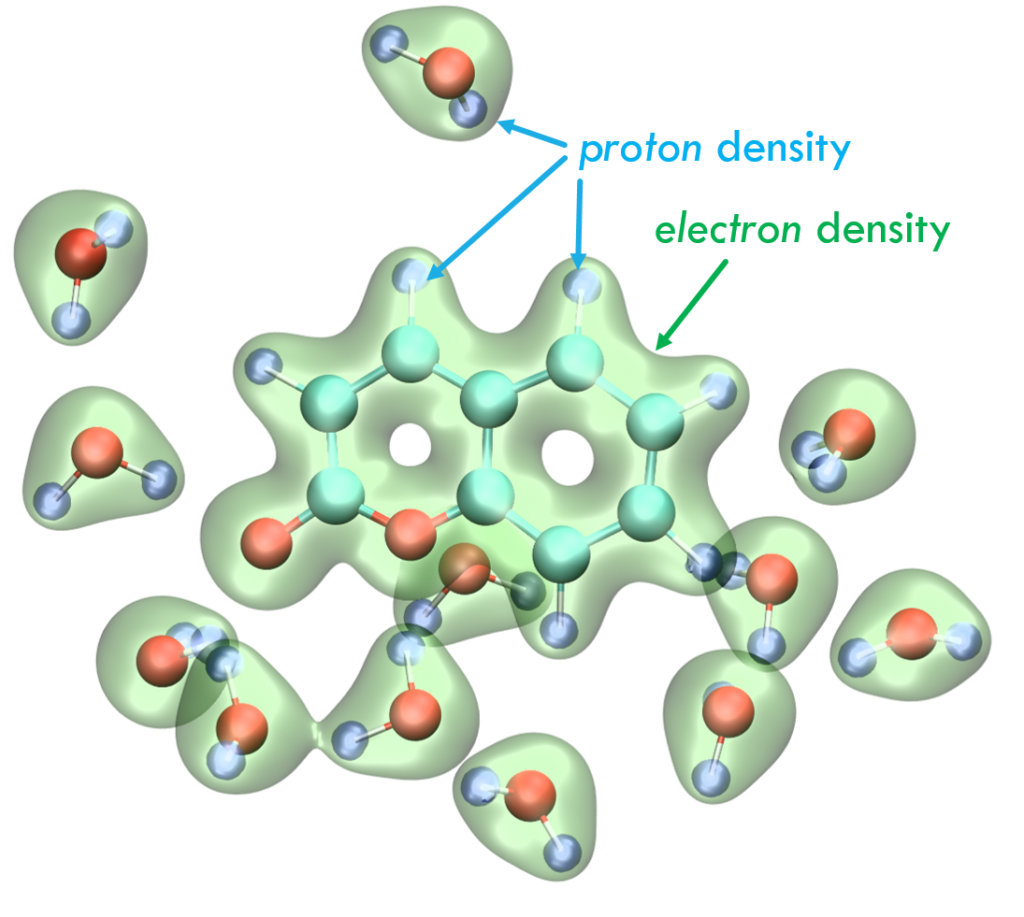
This image shows a deMon2k MDFT calculation of a micro-solvated coumarin molecule. Electrons and protons are treated in an ADFT formalism.
Hybrid QM/MM methods
Hybrid QM/MM methods are one of the indispensable approaches for dealing with complex molecular systems, combining the precision of quantum chemistry with the performance of force fields to handle large-scale systems. deMon2k is equipped with a range of options for carrying out this type of simulation. These include geometry optimization, Born-Oppenheimer dynamics, RT-TDDFT, Ehrenfest MF and multicomponent DFT.
Our team has developed the QIB interface software (QM/MM Input Builder) to prepare input files for deMon2k