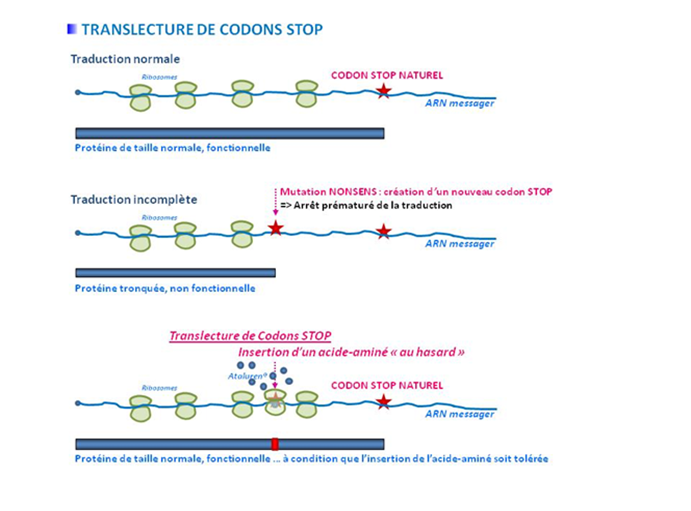
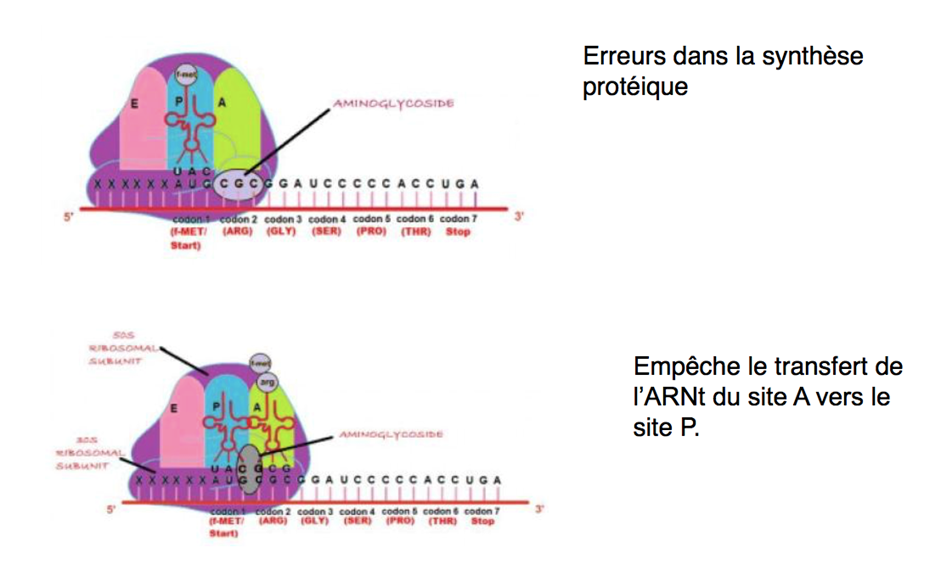
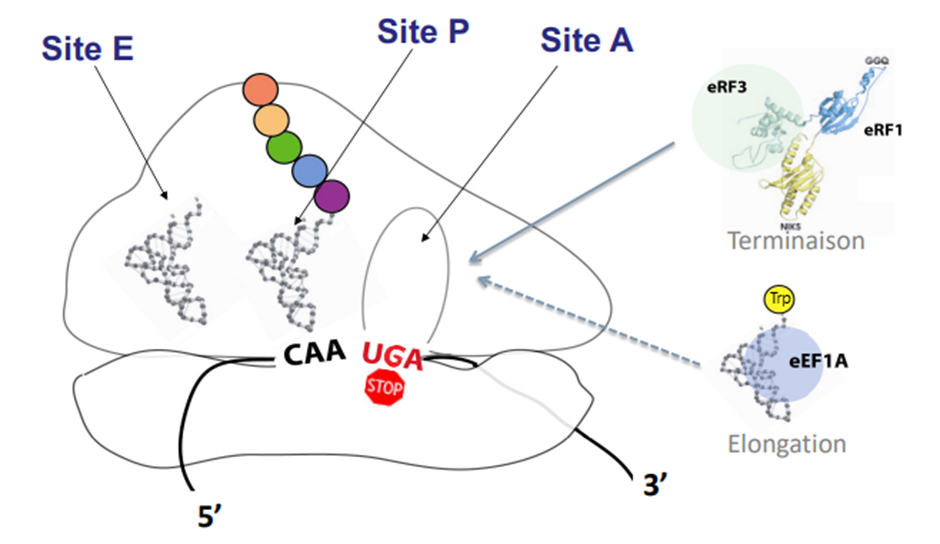
Les codons Stop prématurés (PTC) représentent un tiers des mutations responsable de maladies génétiques et de cancers et sont identifiés chez environ 10% des patients atteints de mucoviscidose.
La mucoviscidose est la maladie génétique grave la plus fréquente dans la population caucasienne.
Dates clés

La mucoviscidose est une maladie rare et génétique qui touche en majorité le système digestif et les voies respiratoires. La mutation la plus fréquente de la mucoviscidose est apparue il y a près de 5 000 dans le sud-ouest de l’Europe.
Voici quelque dates clés qui ont permis une avancée pour la lutte contre cette maladie :
- En 1989 l’équipe de Lap-Chui Tsui découvre que le gène responsable de la maladie est localisé sur le chromosome 7 et qu’il code pour une protéine transmembranaire se nommant Cystic Fibrosis Transmembrane Conductance Regulator (CFTR).
- En 2002 le dépistage néonatal et l’agrément par le ministère de la Santé des Centres de Ressources et de Compétences de la Mucoviscidose a été mis en place au niveau national en France.
CFTR (Cystic fibrosis transmembrane conductance regulator)
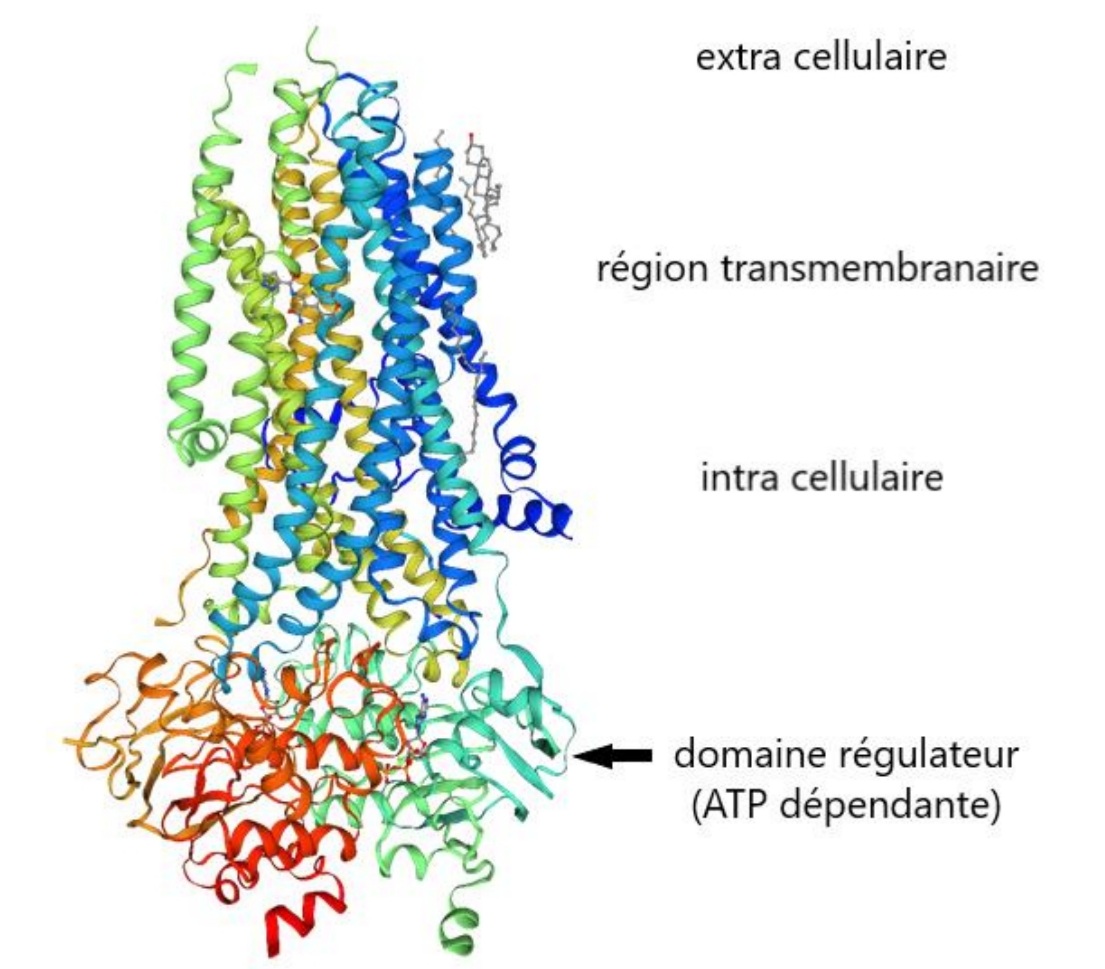
Codée par le gène CFTR situé sur le chromosome 7.
La protéine CFTR fait 1480 acides aminés de taille. C’est une protéine transmembranaire localisée dans la membrane plasmique du pôle apical des cellules épithéliales.
La protéine est un canal ATP dépendante impliqué dans la sécrétion des ions chlorure et joue aussi un rôle dans l’inhibition des canaux d’importation du sodium.
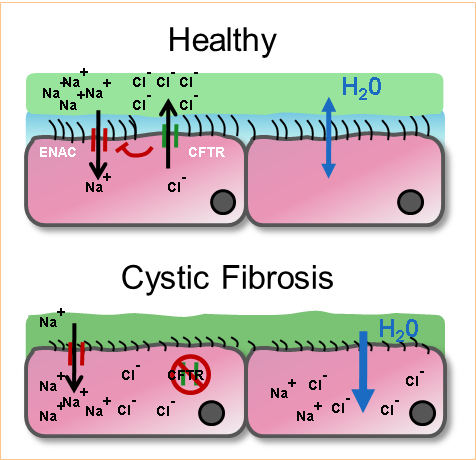
Microbial infection in cystic fibrosis | British Society for Immunology
Si la protéine CFTR est non fonctionnelle les cellules épithéliales seront incapable de sécréter les ions chlorure et d’inhiber l’import sodique, ce qui rend le milieu intra cellulaire fortement saline et par osmose va provoquer la rétention de l’eau de l’espace extracellulaire. La conséquence étant un mucus déshydraté et fortement épais dû à sa viscosité. CFTR étant exprimé de manière constitutive dans les cellules épithéliales, la mucoviscidose touche un grand nombre de tissus comme les voies respiratoires, le pancréas, tube digestif, la peau et aussi l’appareil reproductif.
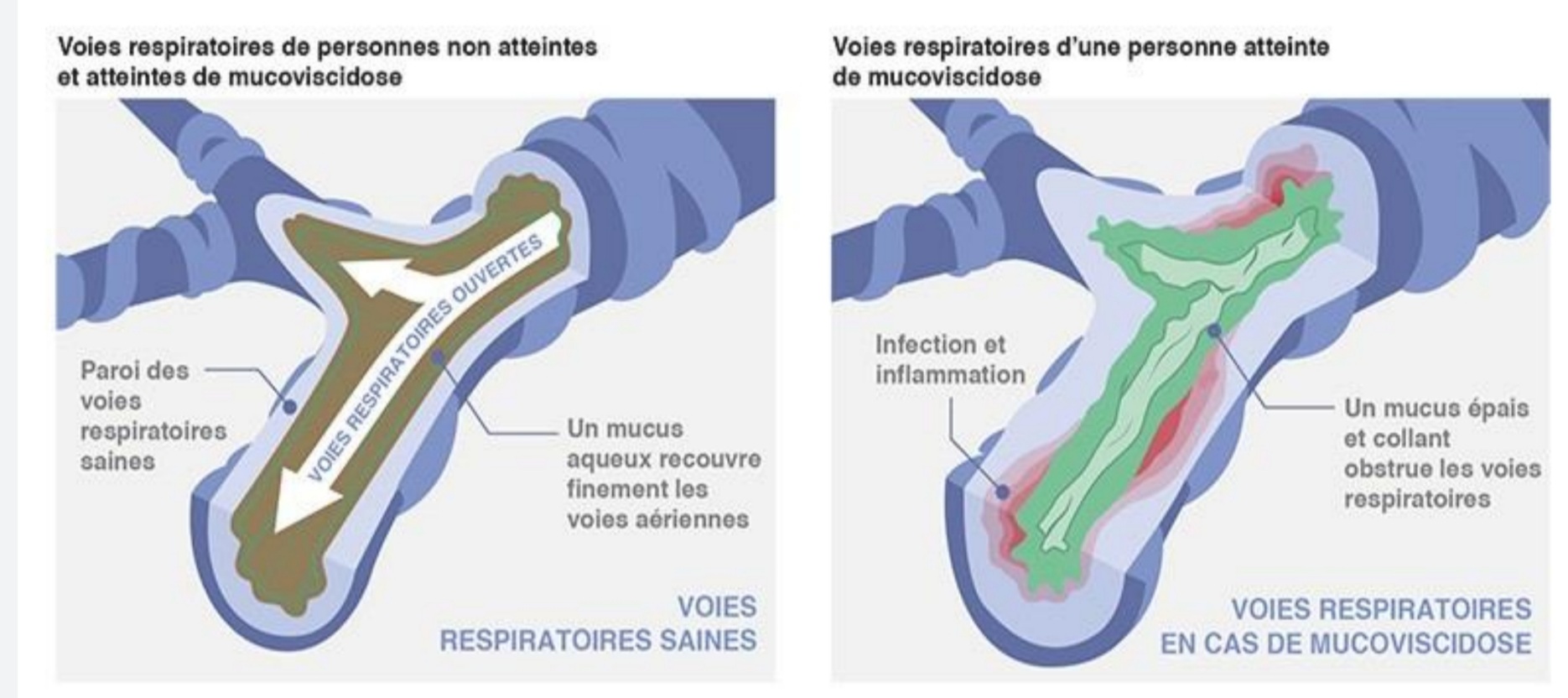
Les symptômes de la Mucoviscidose
Voies respiratoires
L’accumulation de mucus engendre l’inflammation qui s’accompagne de nombreux symptômes :
- Des sifflements, des essoufflements, toux persistante avec des crachats épais
- Infection pulmonaires avec des bactéries comme S.aureus, P.aeruginosa et b. cepacia
- La dilatation des bronches
- Une insuffisance respiratoire qui peut devenir chronique
- Des exacerbations pulmonaires
Prérequis : Des exacerbations sont des périodes d’aggravation des symptômes s’accompagnant d’une diminution de la fonction respiratoire associé à une toux et une augmentation de la production de mucus
Les voies aériennes supérieures
L’atteinte des voies aériennes est récurrente et se traduit par :
- Des sinusites
- Des polypes nasaux (rare) responsables d’une sensation de nez bouché
- Des saignements de nez
Symptôme au niveau digestif et nutritionnel
Le pancréas devant moins fonctionnel entraîne :
- Une altération de la digestion des graisses qui est associé à une diarrhée graisseuse
- Le diabète
Prérequis: une diarrhée graisseuse correspond à une quantité anormalement élevée de lipides dans les selles

- Une dénutrition sévère entraînant une perte de poids et l’ostéoporose
- Intolérance alimentaire à certains composés tel que la protéine de lait de vache,
- Le reflux gastro œsophagien
- L’obstruction des voies biliaires par le mucus épaissi peut causer une stéatose du foie
- Une cirrhose
Prérequis :
Une dénutrition correspond à un apport nutritionnel très faible par rapport au besoin de l’organisme.
L’ostéoporose correspond à une diminution de la densité osseuse ainsi que l’altération de la micro architecture des os
Le reflux gastro œsophagien correspond à une remonté d’une partie du contenu de l’estomac dans l’œsophage
La stéatose du foie correspond à un dépôts graisseux dans le foie
une cirrhose correspond au remplacement progressif des tissus sains par des nodules et tissus fibreux qui altèrent la fonction hépatique
Trouble de la fertilité

- Les hommes atteints de mucoviscidose souffrent d’infertilité dans la plupart des cas car ils présentent un nombre de spermatozoïdes faible, voire nul
- Chez les femmes, la fécondité est un peu diminuée mais la grossesse et le développement du fœtus sont normaux.
Trouble oculaire
Une mauvaise absorption des vitamines (A, D, E et K) peut entraîner une cécité nocturne.
Prérequis: La cécité nocturne est une déficience visuelle qui rend difficile la lecture ou la vision dans la pénombre ou la nuit
Symptôme nouveau né

Les nouveaux nés peuvent présenter :
- Un retard d’émission du méconium qui peut causer une obstruction digestive aiguë de l’intestin .Ce symptôme peut s’accompagner de vomissements ou ballonnements.
- La persistance d’un ictère
- Certains nouveau-nés ont une torsion de l’intestin sur lui-même ou un développement incomplet de l’intestin,
- Faible prise poids entre 4 et 6 semaines chez les bébés
Prérequis : >Le méconium correspond au première selle du nouveau-né de couleur noire. >L’obstruction digestive correspond à un méconium trop épais. >L’ictère correspond à une coloration jaune ou jaunâtre de la peau et du fond de l’œil causé par l’augmentation de la concentration de bilirubine dans le sang
Evolution symptôme et complication
L’évolution de la maladie diffère selon le patient et les organes touchés. Certaines formes apparaissent tardivement et de manière prolongée.
Lors de l’adolescence, et plus encore à l’âge adulte, les symptômes de la mucoviscidose ont tendance à s’accentuer entraînant :
- Chez les adolescents un retard de croissance et une puberté tardive
- La rupture des alvéoles ,l’air circule dans la cavité pleurale (pneumothorax) et affaisse le poumon.
- Hémorragie dans les voies respiratoires
- Une dépression et de l’anxiété,
- Une perte auditive sensorielle
- Bourdonnement d’oreille (acouphènes)
- Trouble du sommeil
- Cancer des canaux biliaires, du pancréas et des intestins
Une prise en charge dès le plus jeune âge et rigoureuse assure au patient une meilleure qualité de vie. Elle permet de diminuer remarquablement la fréquence des symptômes de la mucoviscidose.
Démographie
En France on dénombre environ 6000 patients et 200 enfants naissent avec la mucoviscidose chaque année (1/4500). On s’attend à 9000 patients d’ici 2025.

Sur ce graphique on observe :
- Le nombre de patients passe de 1500 en 1992 à 7000 en 2020 patients suivis. Le gène ayant été découvert en 1989, la maladie était alors récente et peu connue les études et les travaux d’information et de dépistages ont contribué à faire augmenter ce chiffre.
- La proportion adulte/enfant s’inverse. En effet, on passe de 17.8% d’adultes suivis et traités en 1992 à 59.7% en 2020,cela signifie que l’espérance de vie a augmenté.
La deuxième hypothèse se confirme en observant la pyramide des âges :

En 10 ans on observe un élargissement de la pyramide des âges. Cela signifie que ces 10 dernières années, l’espérance de vie a augmenté. Les couleurs foncées représentent les données de 2020 et les données claires sont celles enregistrées en 2010.
On observe qu’en 2020 il y a 120 garçons dans la population française atteints de mucoviscidose. Il y en avait 75 en 2010. L’écart s’intensifie lorsque l’on monte dans la pyramide.
On avait environ 35 femmes de 31 ans atteintes de mucoviscidose il y a 12 ans tandis qu’on on dénombrait environ 85 il y a 2 ans.
On observe également le rétrécissement des effectifs après l’espérance de vie moyenne (50 ans).

On observe ce type de pyramide pour la population française générale. On conclut que le nombre d’enfants qui naissent avec la maladie diminue et l’espérance de vie se rallonge.
Géographie

On observe de très fortes disparités de l’incidence au niveau régional.
Le Nord-Pas-de-Calais, la Bretagne et la Provence-Alpes-Côte-d’Azur ont des incidences qui peuvent atteindre 18 malades pour 100 000 habitants. Tandis que des départements comme l’Essonne ont des incidences à 8 malades pour 100 000 habitants.
Cela représente 1 naissance sur 3000 en Bretagne et 1 naissance sur 7800 dans le Languedoc-Roussillon (1/4500 étant la moyenne nationale).
Mortalité
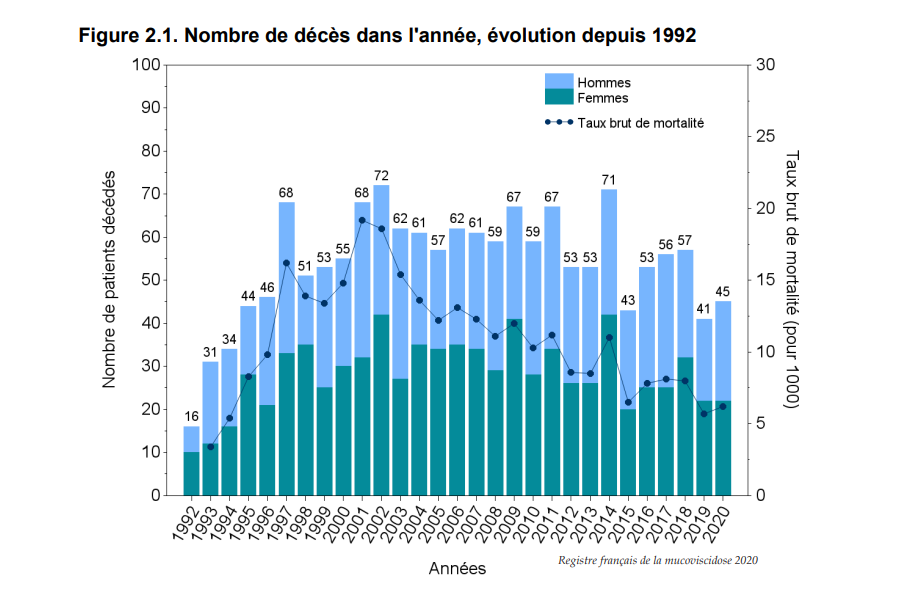
En ce qui concerne la mortalité, On observe entre les années 1992 et 2002 une hausse des décès et du taux brut de mortalité (En démographie, le taux de mortalité (ou taux brut de mortalité) est le rapport entre le nombre annuel de décès et la population totale moyenne sur une période et dans un territoire donné.) qui touche de manière égale les hommes et les femmes.
Depuis 2002 en revanche le taux brut de mortalité est en très nette hausse tandis que le nombre de patients décédés est quasi stagnant. Il ne faut pas oublier que le nombre de patients est en hausse et que ces chiffres sont très encourageants.
Autres chiffres mesurables

Le nombre de transplantations dans l’année est également en nette hausse. I
Dépistage

Depuis 2002, la mucoviscidose fait parties des 6 maladies qui sont dépister à la naissance.
Le diagnostic prénatal de la mucoviscidose consiste à prélever du liquide amniotique au cours d’une amniocentèse ou des cellules du placenta dans l’utérus de la femme enceinte, afin de réaliser une analyse génétique de l’embryon
Un dépistage néonatal (test de Guthrie), Effectué au troisième jour de la vie, le dépistage prend la forme d’une prise de sang fait au niveau du talon du nouveau-né. Cet examen sanguin permet de réaliser la dosage d’une enzyme fabriqué par la pancréas, la trypsine immunoreactive ou TIR
Si les analyses révèlent un taux élevé de la trypsine un test génétique qui consiste à étudier le gène de la protéine CFTR est fait. La recherche s’effectue sur les 30 mutations les plus fréquentes
Cet examen permet d’identifier un ou deux mutations de ce dernier.
Si les examens révèlent un ou deux mutations de gènes ou si l’hypertrypsinemie( taux élevé de la trypsine immunoreactive) persiste, un test de sueur est proposé.ce dernier donne la possibilité de faire un diagnostic.
En accord avec la législation française, unconsentement parental écrit au dos du carton est demandé pour tout nouveau-né et permettra de réaliser l’analyse des mutations CFTR si la valeur de la TIR est au-dessus du seuil (0,5 % des nouveau-nés).
En cas de non consentement parental
(absence ou refus) ou si aucune mutation n’est retrouvée avec une valeur de TIR J3 ultra-haute(, un contrôle de TIR est réalisé vers le 21e jour (J21).
les enfants chez qui on a identifié deux ou une mutation(s) ou une TIR J21 au-dessus du seuil sont convoqués, afin de réaliser un test de la sueur (TS) qui consiste à stimuler la transpiration au niveau de l’avant bras en utilisant un courant électrique de faible intensité avant de recueillir la sueur et d’analyser. Une concentration de chlore supérieur à 60millimoles par litres permet de dire que la personne est malade.) qui réfutera ou confirmera le diagnostic de MV.
au 3ème jour de vie. Si ce premier dosage révèle une valeur supérieure à un seuil d’alerte de 55 µg/l (dosage appelé TIR1 à J3), alors deux nouveaux dosages au 3ème jour (dosage appelé TIRmoy à J3) sont réalisés, et si leur moyenne est supérieure ou égale à un seuil d’action de 65 µg/l,
alors une recherche des mutations du gène CFTR est effectuée.