J. D Samaniego-Rojas, L. I. Hernández-Segura, L. López-Sosa, R. I Delgado-Venegas, B. Gomez, J.-C. Lambry, A. de la Lande, T. Mineva, J. Alejandre, B. A Zúñiga-Gutiérrez, R. Flores-Moreno, P. Calaminici, G. Geudtner, A. M Köster.Andreas in Multiscale Dynamics Simulations: Nano and Nano-bio Systems in Complex Environments.Edited by D. Wei and D. R. Salahub. 2021. Royal Chemical Society. doi.org/10.1039/9781839164668-00001
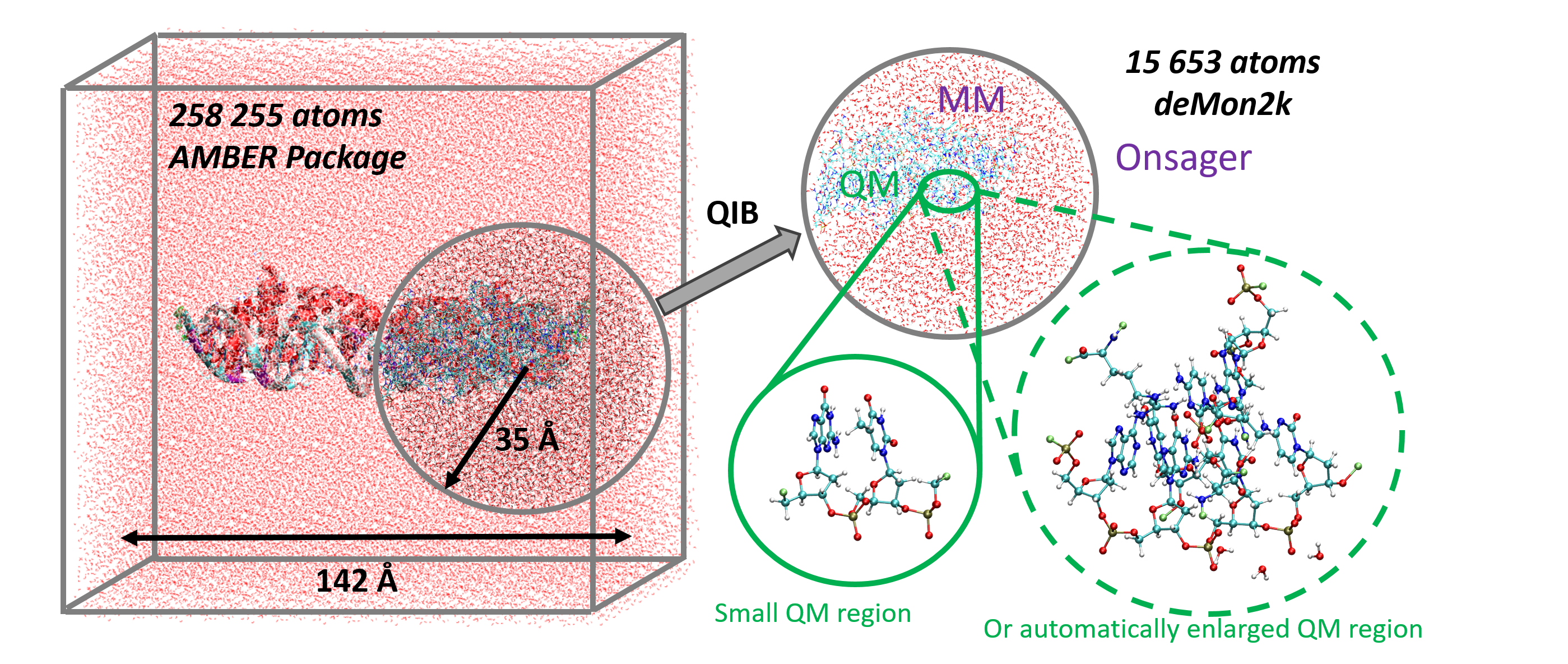
This chapter describes the theoretical background of the quantum mechanical/molecular mechanical (QM/MM) implementation in deMon2k within the framework of auxiliary density functional theory (ADFT). It aims to give the reader an overview of the current state of the art of this QM/MM implementation and perspectives for its future development. To this end, we first derive the ADFT working equations for the QM and QM/MM energy and gradient expressions. Based on the joint QM/MM gradient expression, we present algorithms for QM/MM structure optimizations, transition-state searches and molecular dynamics simulations. The use of auxiliary density perturbation theory (ADPT) in the framework of QM/MM is discussed using illustrative implementations including analytic second-order ADFT energy derivatives, nuclear magnetic resonance chemical shift calculations and excited state calculations using time-dependent ADFT. The chapter closes with the description of a transformation program used to generate deMon2k QM/MM inputs.