Pablo Antonio Martínez, Theresa Vock, Liliane Racha Kharchi, Jesus Nain Pedroza-Montero, Xiaojing Wu, Karim Hasnaoui, Aurélien de la Lande. Comput. Phys. Comm. 2023, 108946, in press.https://doi.org/10.1016/j.cpc.2023.108946
This article belongs to Special Issue on Attosecond Chemistry software in Computer Physics Communications
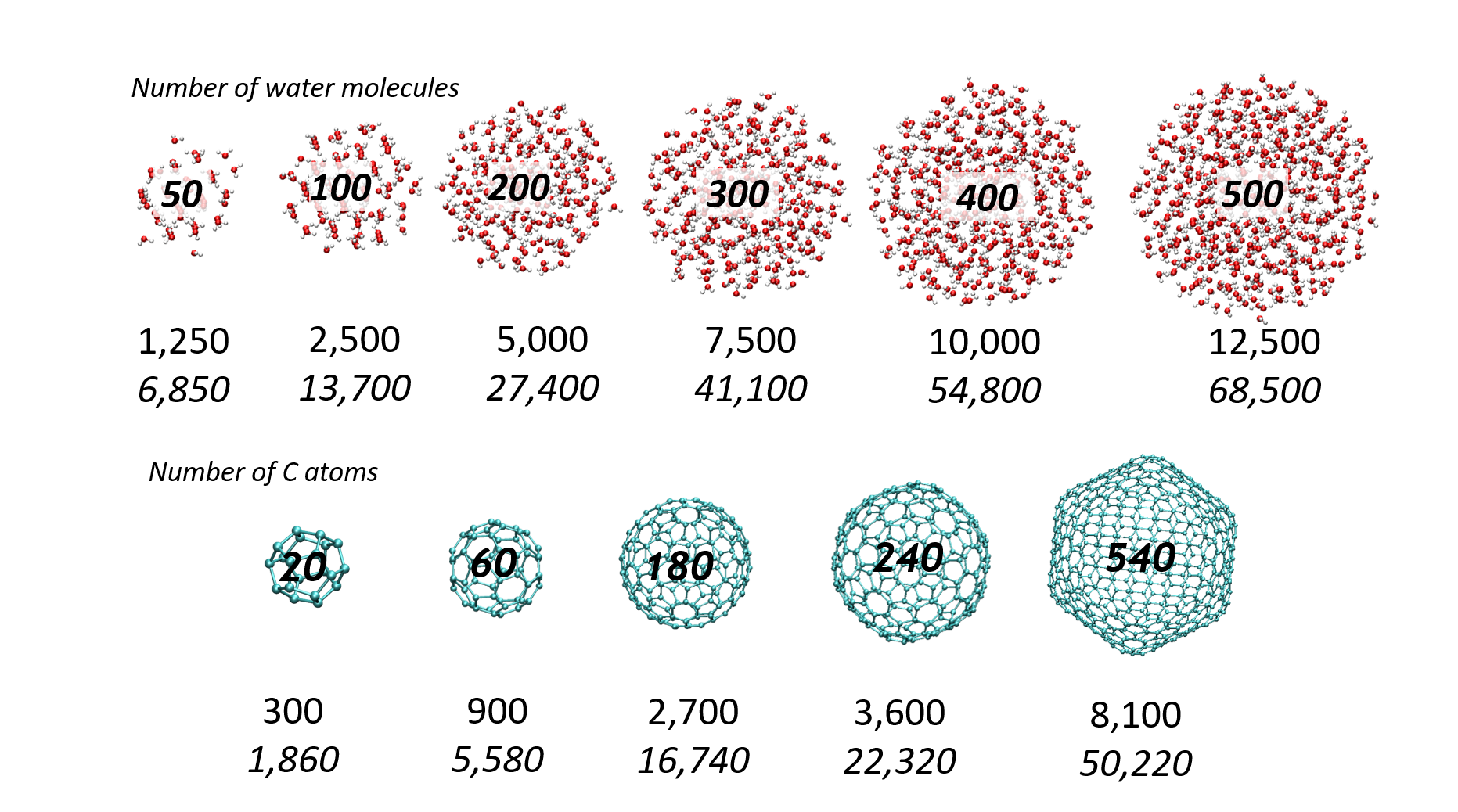
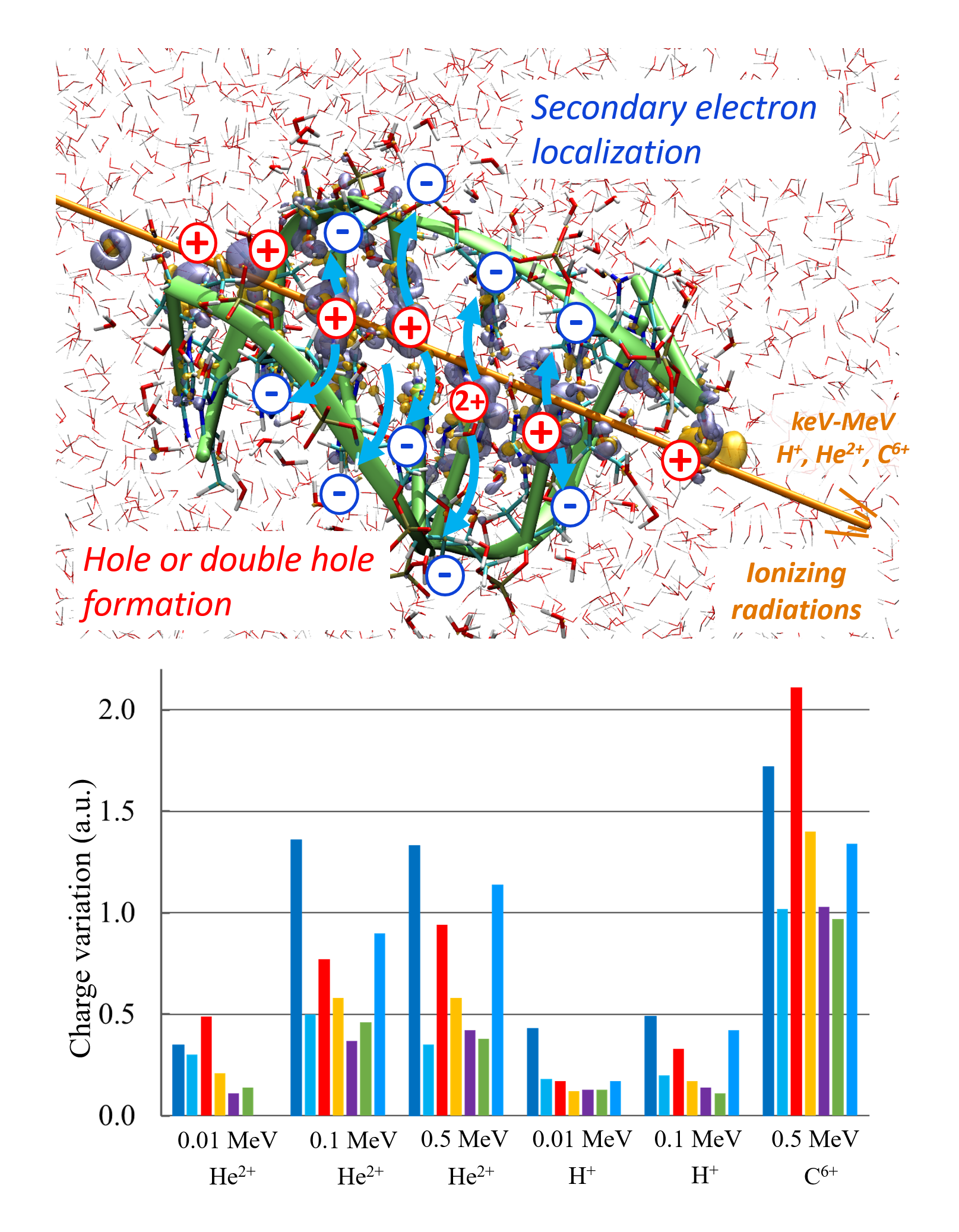
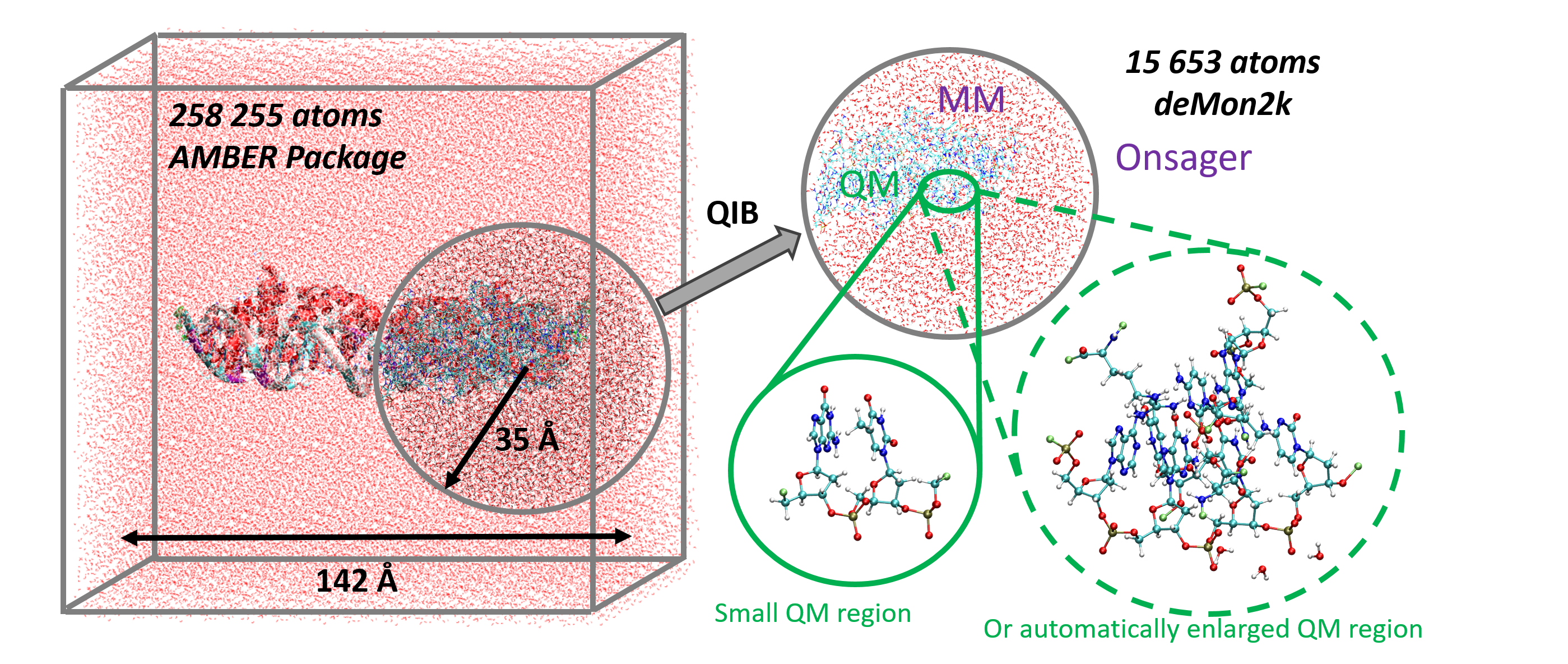
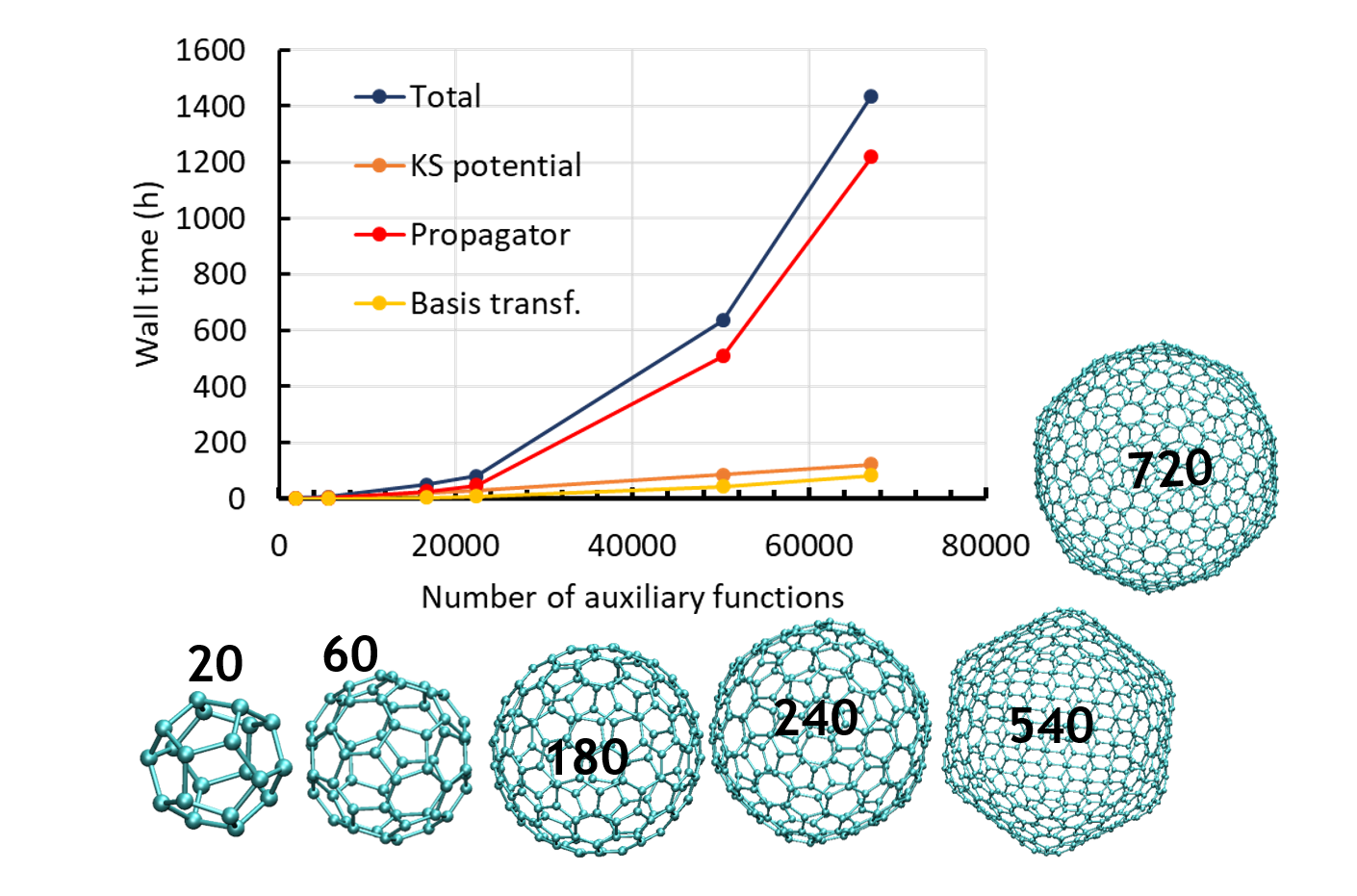
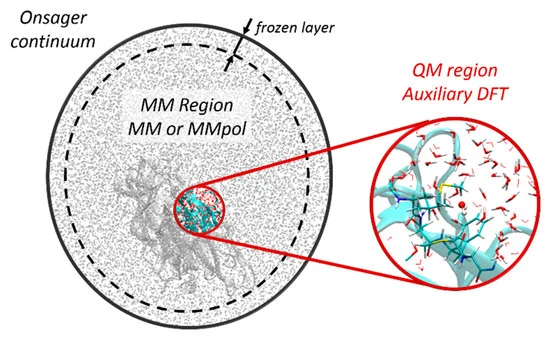
We report a new Multi-GPU (Graphical Processor Unit) implementation of real-time time-dependent Auxiliary Density Functional Theory (DFT) for simulations of attosecond electronic dynamics in molecular systems subjected to strong perturbations. Our code relies on the Kohn-Sham formalism of DFT and has been implemented in the deMon2k Fortran code. We expand single-particle wave functions (i.e molecular orbitals) as linear combinations of Gaussian-type-orbitals centered on atoms. The density matrix propagation is carried out on GPU while the Kohn-Sham potential is operated on CPUs (Central Processor Unit) with the help of variationally fitted densities. We propose a parallelization strategy using the MAGMA/CUDA libraries to calculate the exponential of dense Hermitian matrices entering the mathematical definition of the propagator, here using Taylor expansions. We report performance benchmarks on water droplets and on fullerenes (C50 to C540). They show a clear advantage of GPU over CPU (using the Scalapack library). The benchmarks also show the benefit of using more than one GPU for systems comprised of up to more than 10,000 basis functions. There, a speed-up of almost 40 between pure 40 CPU and four 4 GPU is obtained. Attosecond electron dynamics simulation in molecular systems comprised of several thousands of electrons becomes amenable to routine simulations in our code. We assess the accuracy of the GPU implementation considering various applications, namely, the calculation of extreme UV absorption spectra with non-Hermitian dynamics, the response of C180 to an electric perturbation, and finally the irradiation of a DNA/protein complex by a 0.4 MeV proton. The results demonstrate the robustness of the implementation. This work also paves the way for future even more efficient implementations.