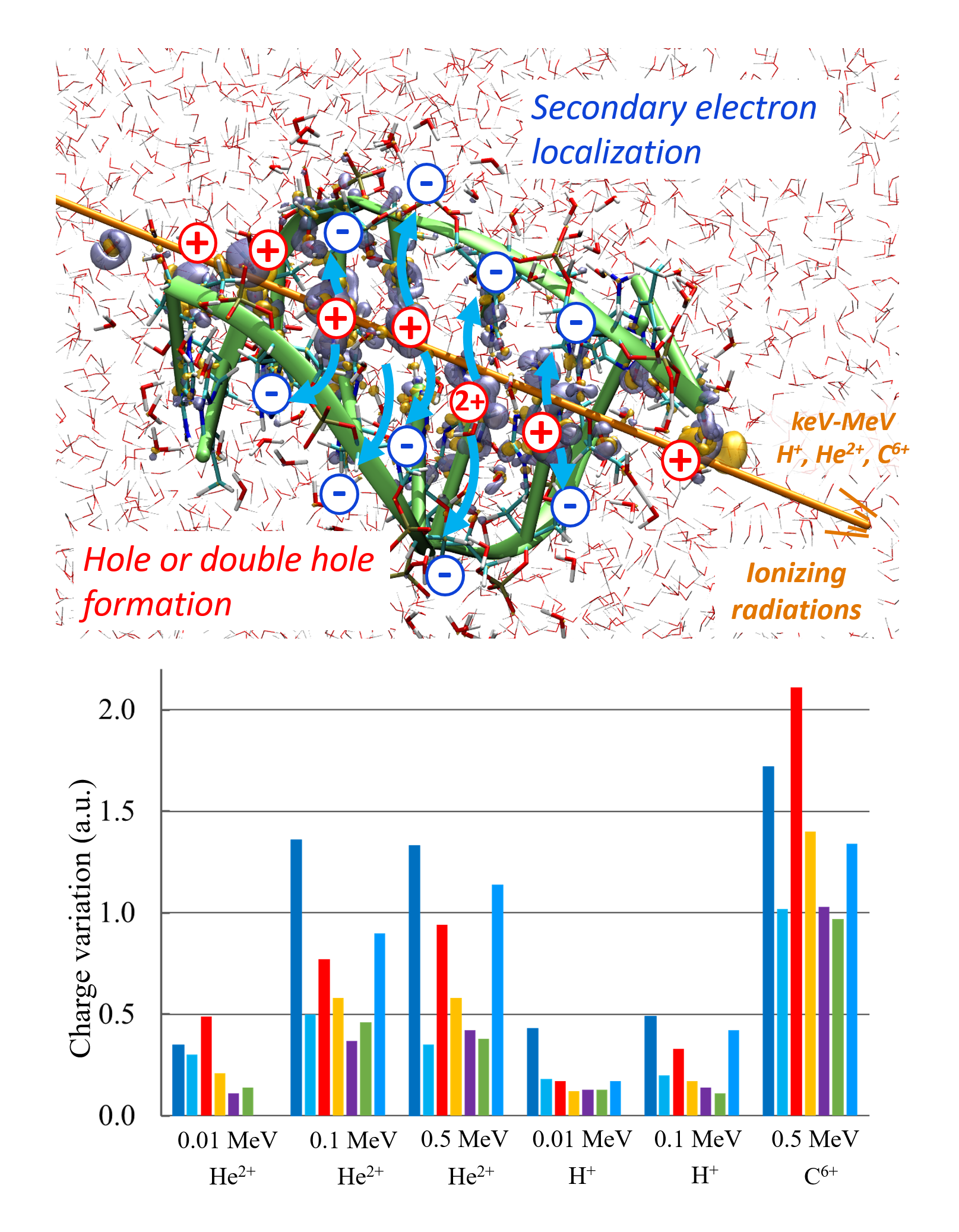
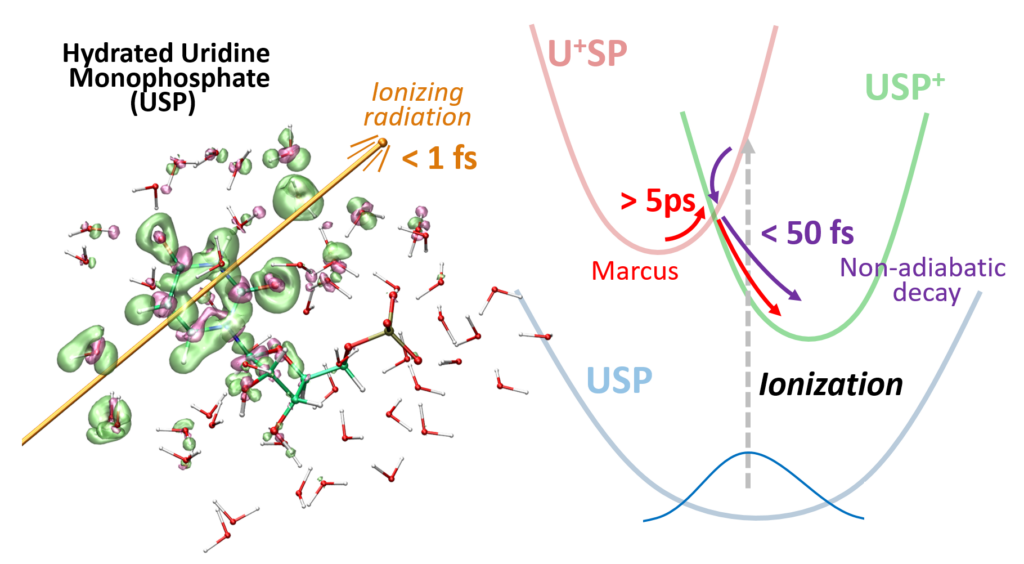
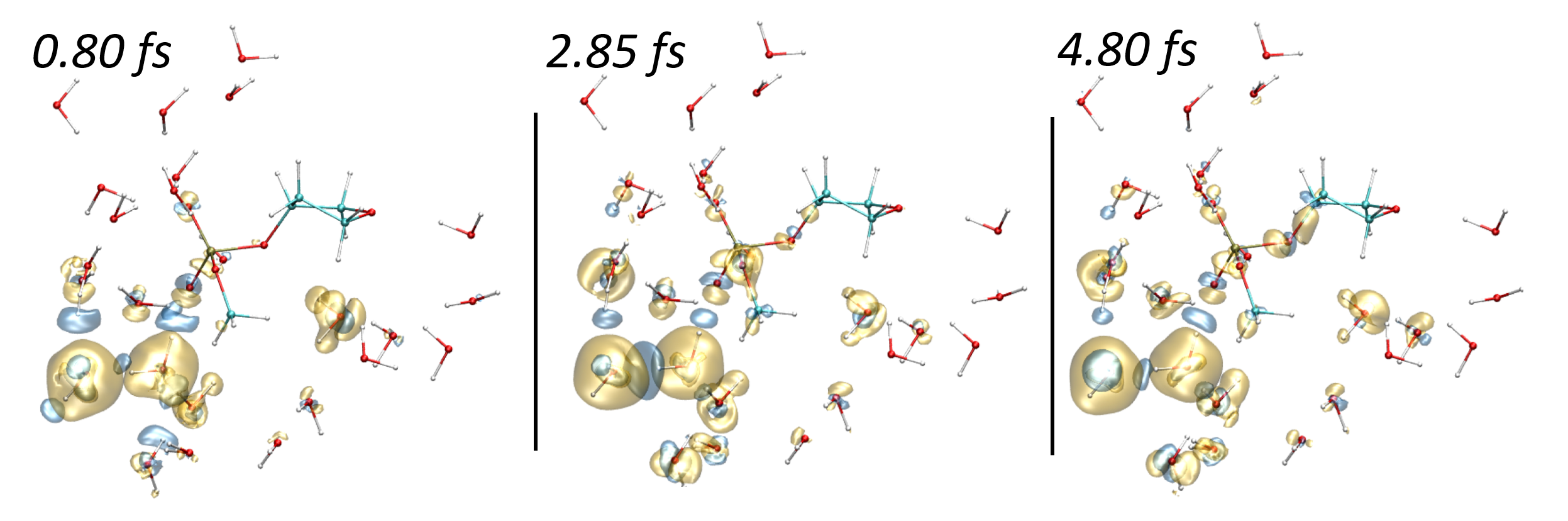
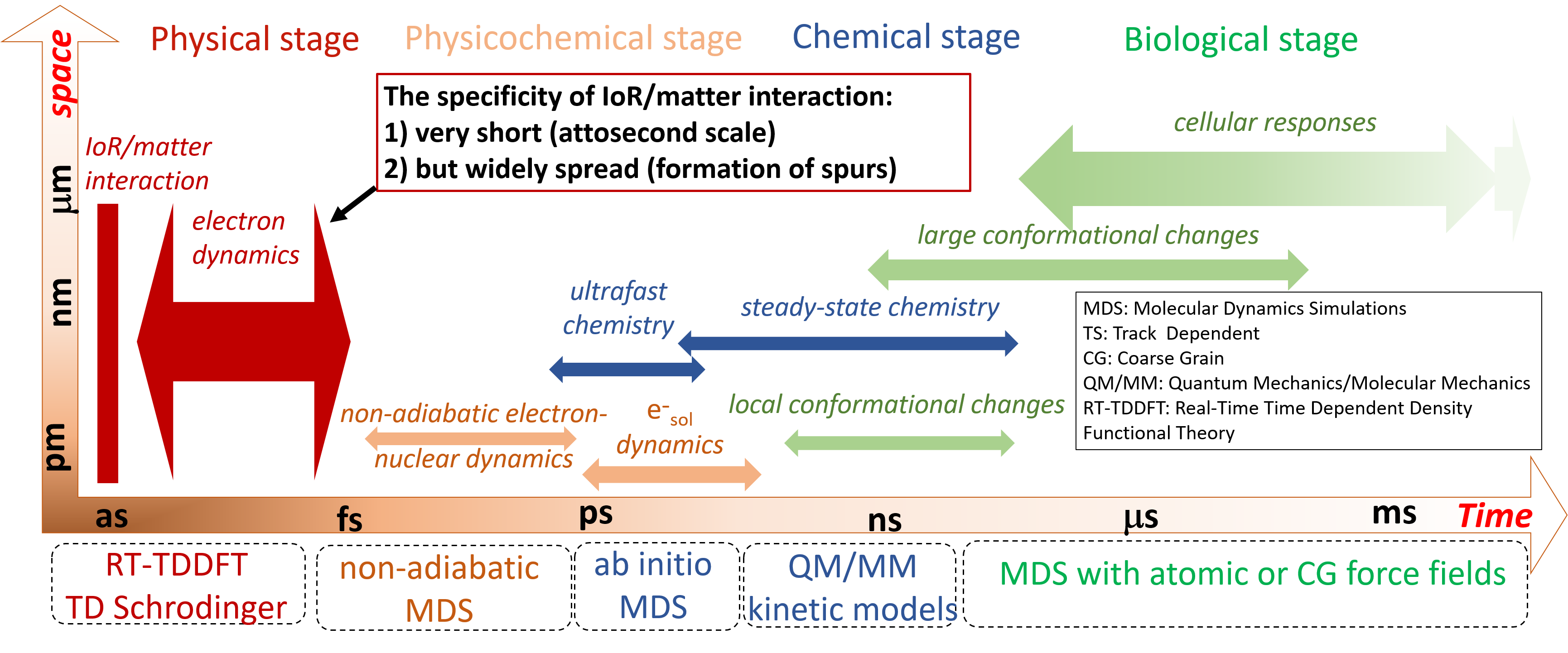
Damien Tolu, Dominique Guillaumont, Aurélien de la Lande. J. Phys. Chem. A. 2023, 127, 34, 7045–7057. doi.org/10.1021/acs.jpca.3c02117. Link to full text in HAL.

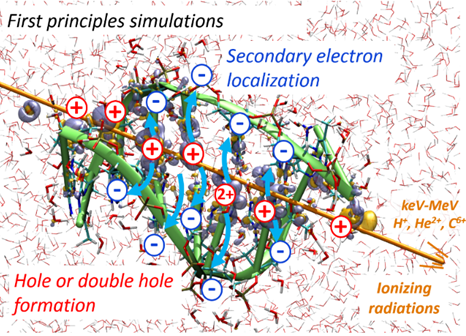
The PUREX solvent extraction process, widely used for recovering uranium and plutonium from spent nuclear fuel, utilizes an organic solvent composed of tributyl phosphate (TBP). The emission of ionizing particles such as alpha particles, resulting from the decay of plutonium, makes the organic solvent vulnerable to degradation. Here, we study the ultrashort time alpha irradiation of tributylphosphate (TBP) and Pu(NO3)4(TBP)2 complex formed in the PUREX process. Electron dynamics is propagated by Real-Time-Dependent Auxiliary Density Functional Theory (RT-TD-ADFT). We investigate the use of previously proposed absorption boundary conditions (ABC) in the molecular orbital space to treat secondary electron emission. Basis set and exchange correlation functional effects with ABC are reported as well as a detailed analysis of the ABC parametrization. Preliminary results on the water molecule and then on TBP show that the phenomenological nature of the ABC parameters necessitates selecting appropriate values for each system under study. Irradiation of free and complexed TBP shows an influence of the ligands on the variation of atomic charges on the femtosecond time scale. An accumulation of atomic charges in the alkyl chains of TBP is observed in the case where the nitrate groups are predominantly irradiated. In addition, we find that the Pu atom regains its electric charge very rapidly after being hit by the projectile, with the coordination sphere serving as an electron reservoir to preserve its formal redox state. This study paves the road toward a full understanding of the degradation of organic extracants employed in the nuclear industry.