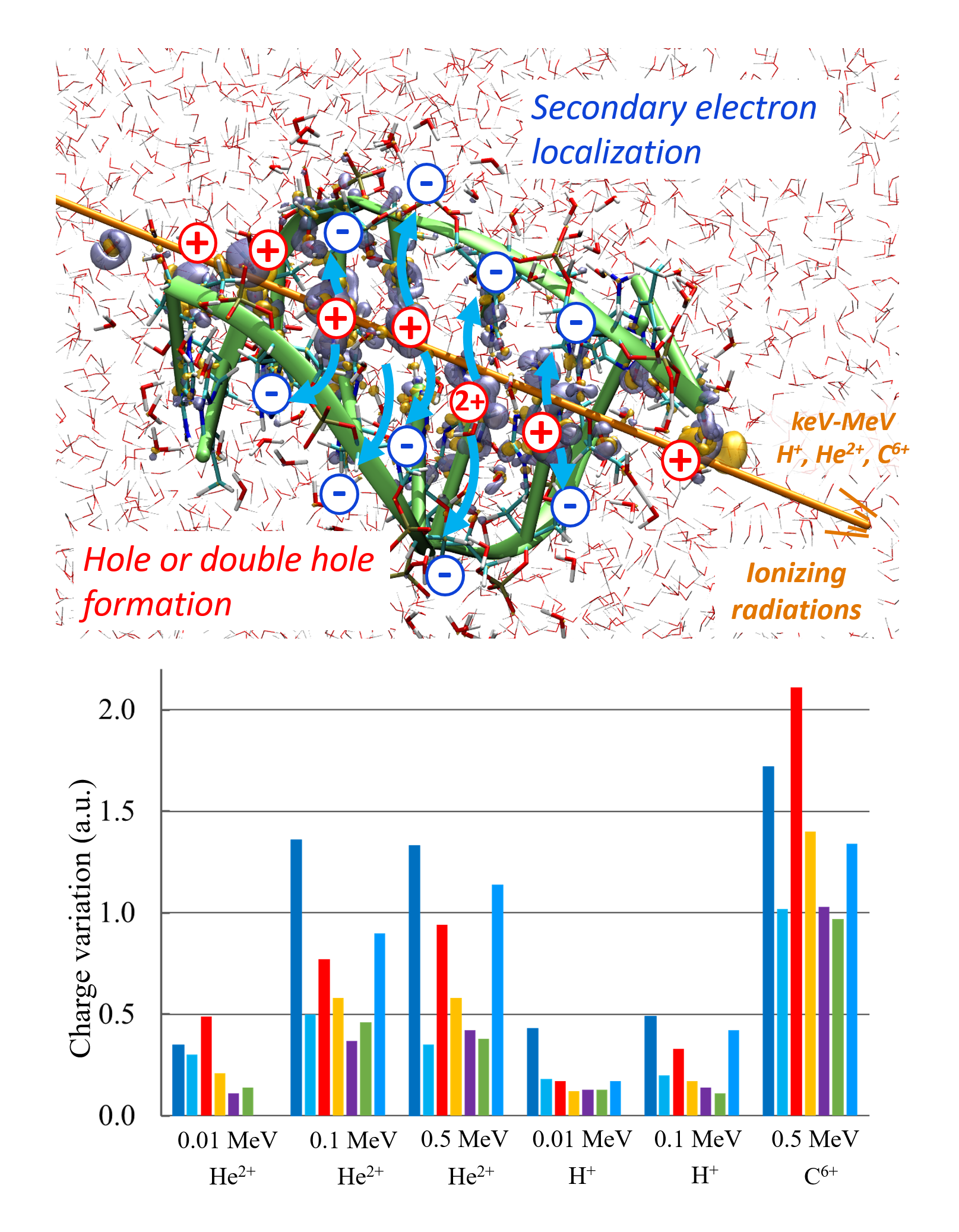
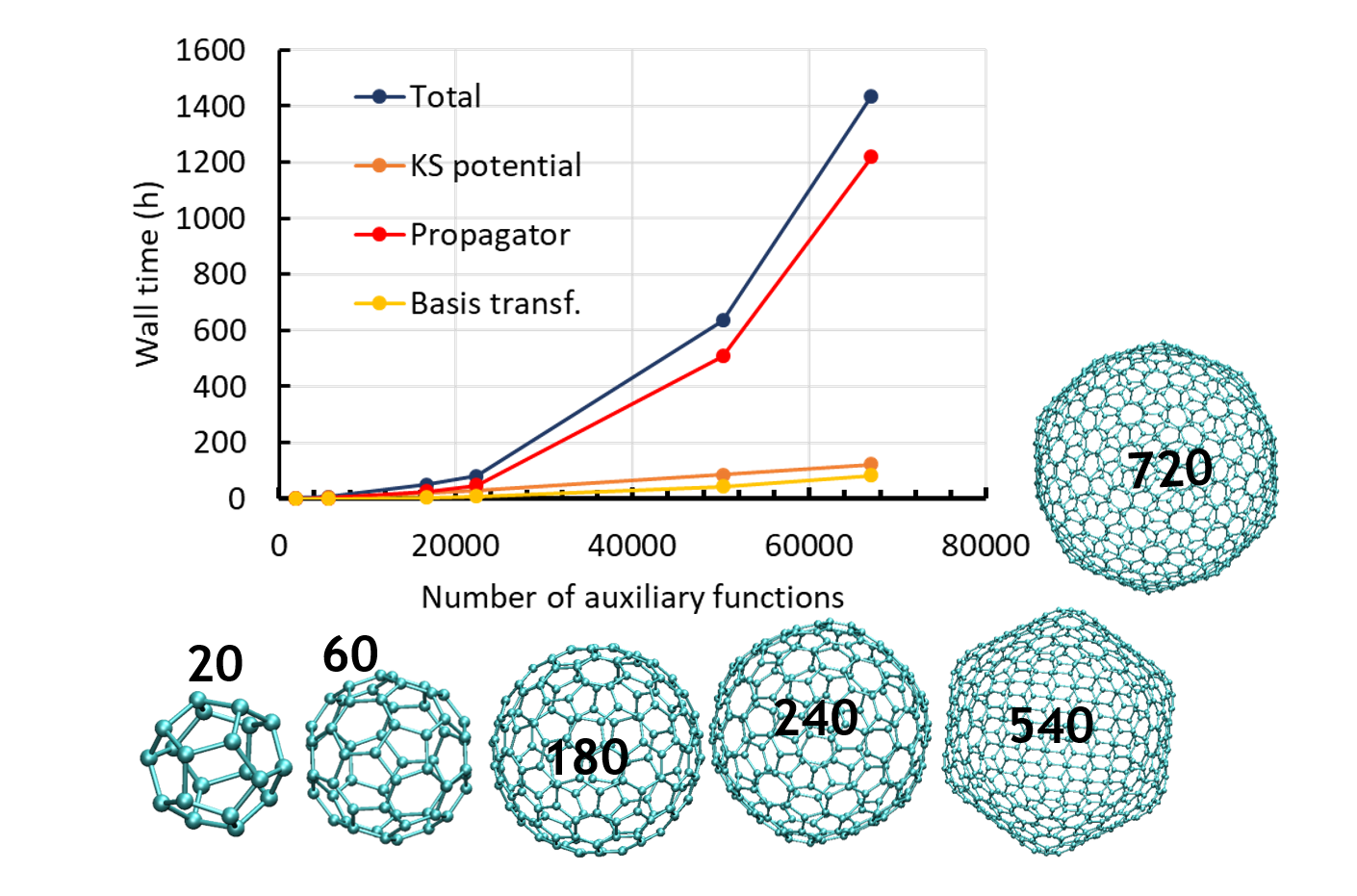
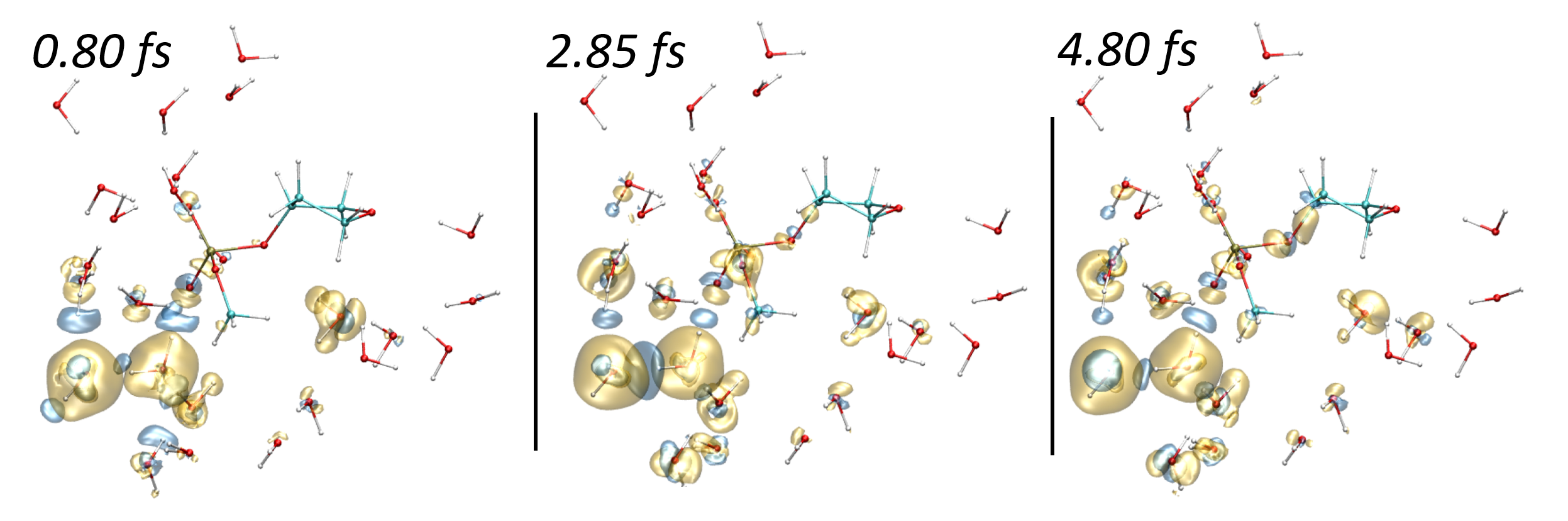
Rika Tandiana, Karwan Ali Omar, Eleonora Luppi, Fabien Cailliez, Nguyen-Thi Van-Oanh, Carine Clavaguéra, Aurélien de la Lande. J. Chem. Theor. Comput. 2023, 19, 21, 7740–7752. doi.org/10.1021/acs.jctc.3c00656. Link for full text in HAL.

The electronic stopping power is an observable property that quantifies the ability of swift ions to penetrate matter to transfer energy to the electron cloud. The recent literature has proven the value of Real-Time Time-Dependent Density Functional Theory to accurately evaluate this property from first-principles, but questions remain regarding the capability of computer codes relying on atom-centered basis functions to capture the physics at play. In this Perspective, we draw attention to the fact that irradiation by swift ions triggers electron emission into the continuum, especially at the Bragg peak. We investigate the ability of Gaussian atomic orbitals (AOC), which were fitted to mimic continuum wave functions, to improve electronic stopping power predictions. AOC are added to standard correlation-consistent basis sets or STO minimal basis sets. Our benchmarks for water irradiation by fast protons clearly advocate for the use of AOC, especially near the Bragg peak. We show that AOC only need to be placed on the molecules struck by the ion. The number of AOC that are added to the usual basis set is relatively small compared to the total number of atomic orbitals, making the use of such a basis set an excellent choice from a computational cost point of view. The optimum basis set combination is applied for the calculation of the stopping power of a proton in water with encouraging agreement with experimental data.