Karwan A Omar, Feven A Korsaye, Rika Tandiana, Damien Tolu, Jean Deviers, Xiaojing Wu, Angela Parise, Aurelio Alvarez-Ibarra, Felix Moncada, Jesus Nain Pedroza-Montero, Daniel Mejía-Rodriguez, Nguyen-Thi Van-Oanh, Fabien Cailliez, Carine Clavaguéra, Karim Hasnaoui, Aurélien de la Lande. Eur. J. Special Topics, 2023. doi.org/10.1140/epjs/s11734-023-00905-6. Full text in HAL.
Special collection: Ultrafast Phenomena from attosecond to picosecond timescales: theory and experiments
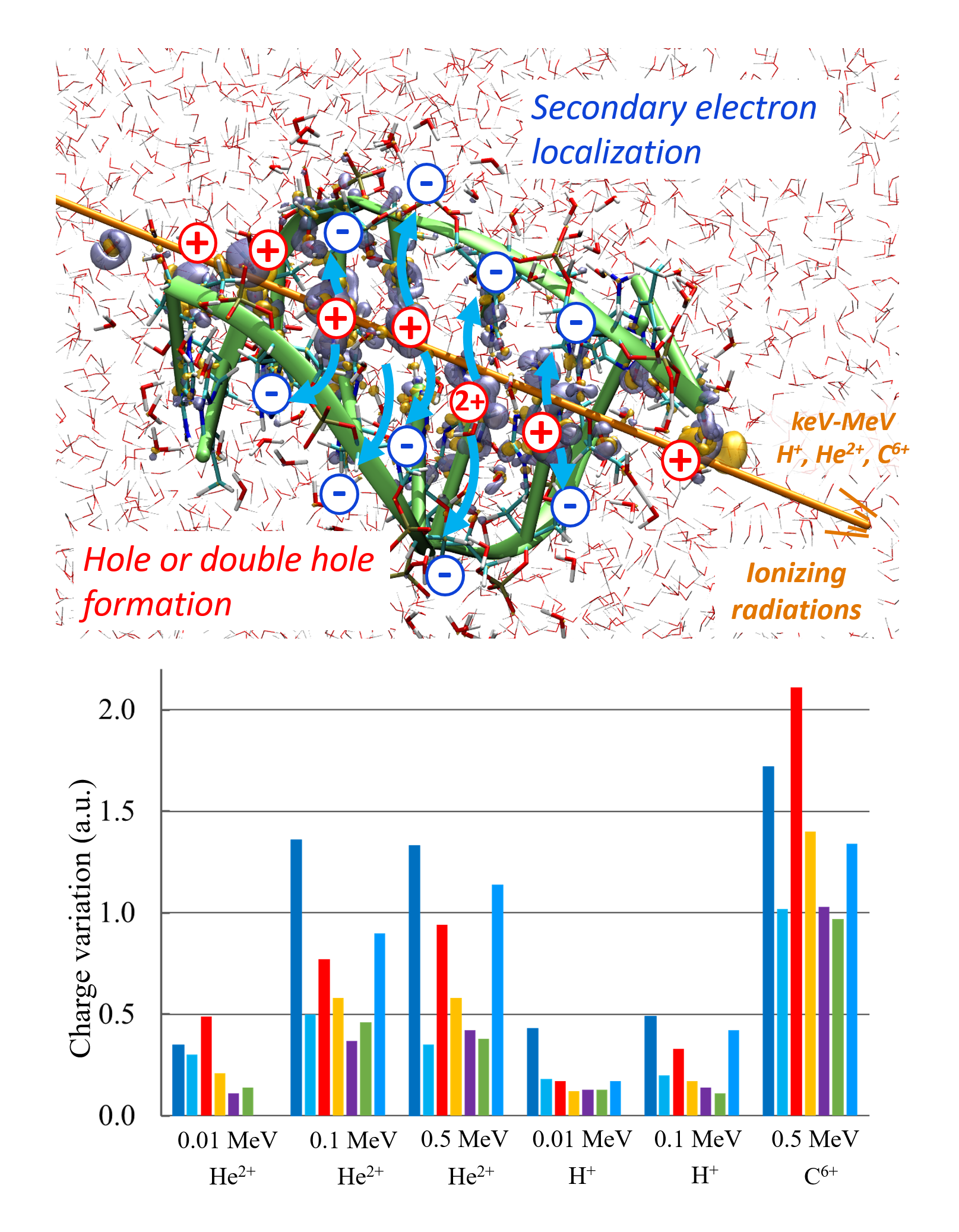
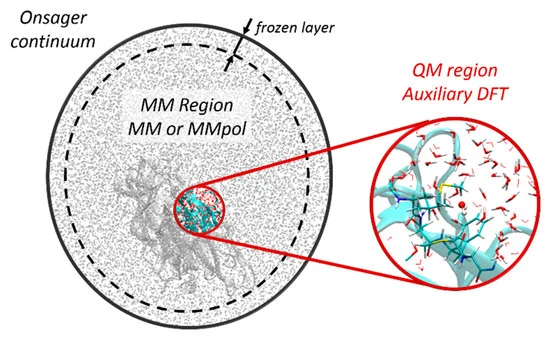
We summarize in this article the recent progress made in our laboratories in the development of numerical approaches dedicated to investigating ultrafast physicochemical responses of biological matter subjected to ionizing radiations. Our modules are integrated into the deMon2k software which is a readily available program with highly optimized algorithms for conducting Auxiliary Density Functional Theory (ADFT) calculations. We have developed a computational framework based on Real-Time Time-dependent ADFT to simulate the electronic responses of molecular systems to strong perturbations, while molecular dynamics simulations in the ground and excited states (Ehrenfest dynamics) are available to simulate irradiation-induced ultrafast bond breaking/formation. Constrained ADFT and Multi-component ADFT have also been incorporated to simulate charge transfer processes and nuclear quantum effects, respectively. Finally, a coupling to polarizable force fields further permits to realistically account for the electrostatic effects that the systems’ environment has on the perturbed electron density. The code runs on CPU or hybrid CPU/GPU architectures affording simulations of systems comprised up to 1000 atoms at the DFT level with controlled numerical accuracy. We illustrate the applications of these methodologies by taking results from our recent articles that aimed principally at understanding experimental data from pulse radiolysis experiments.