Rika Tandiana, Carine Clavaguera, Karim Hasnaoui, Jesús Naín Pedroza-Montero, Aurélien de La Lande. Theor. Chem. Acc. 2021, 140, 126. doi.org/10.1007/s00214-021-02819-9. Full text in HAL.
Part of a collection: 20th deMon Developers Workshop
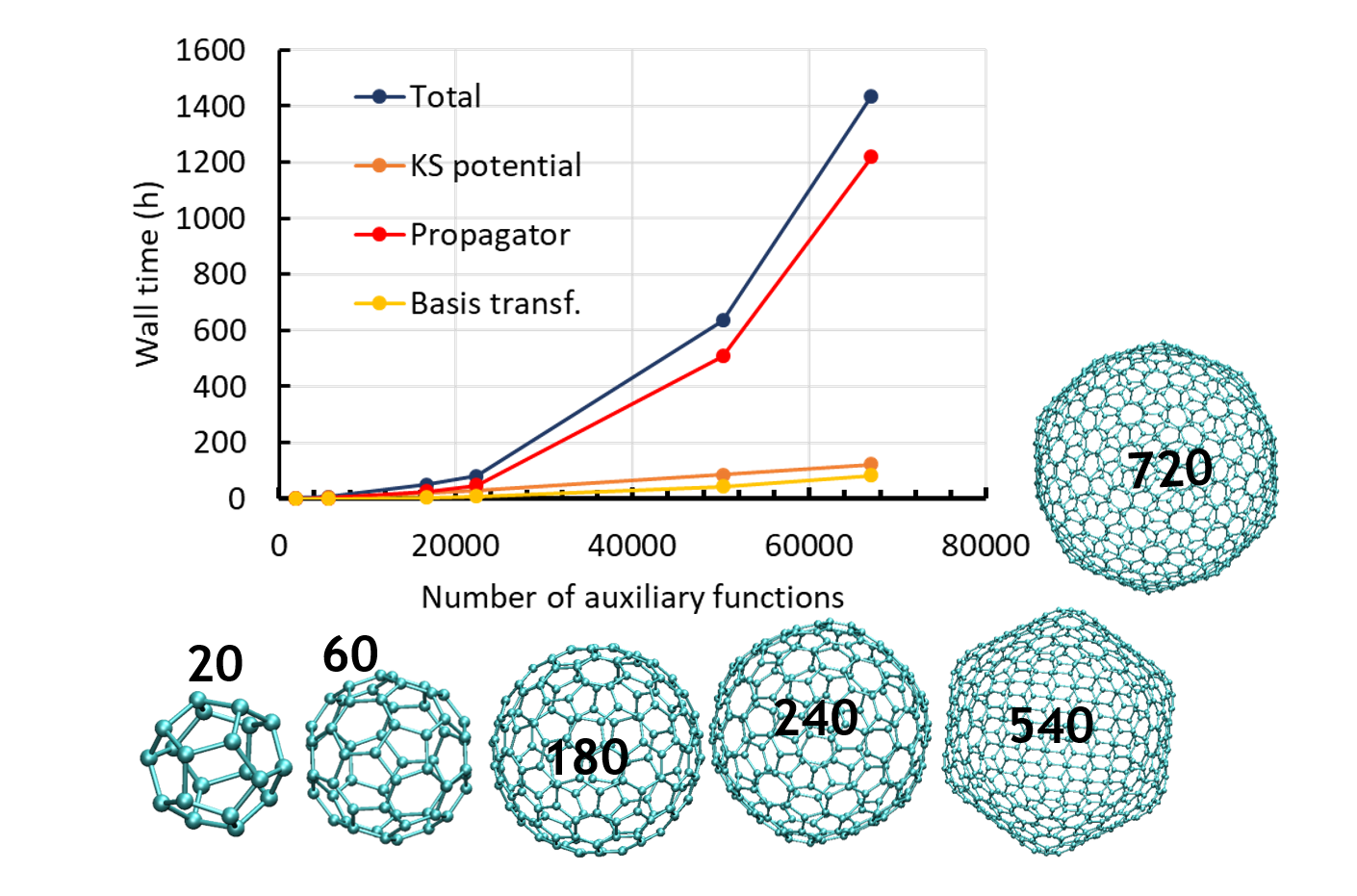
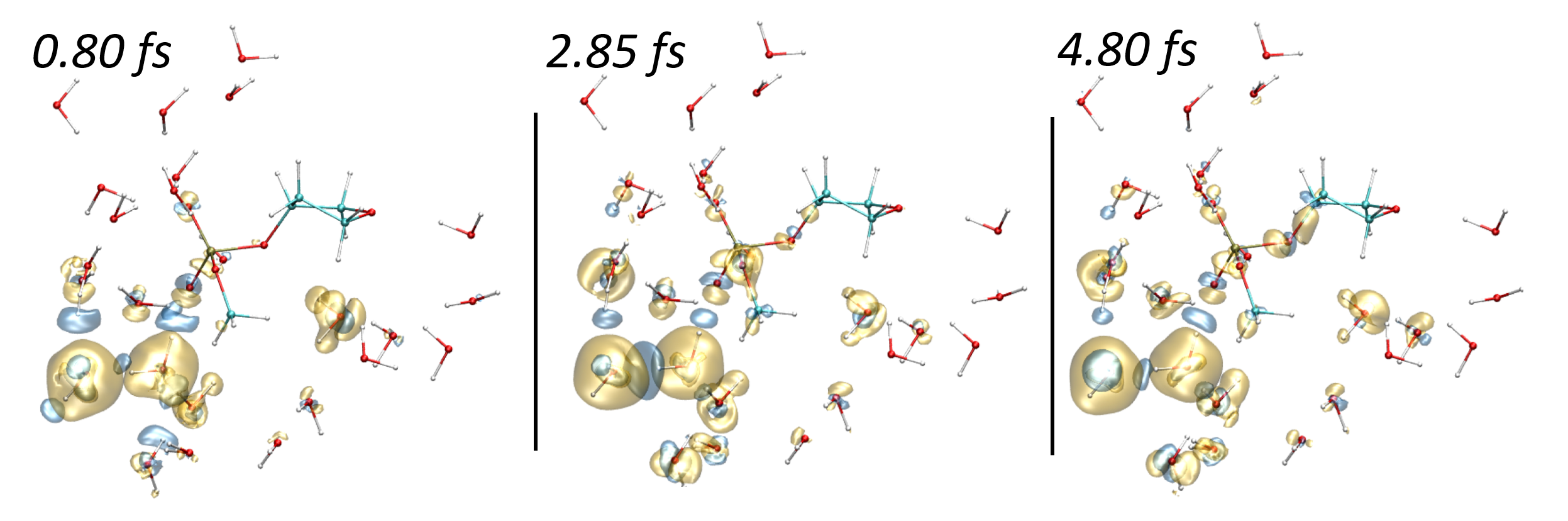
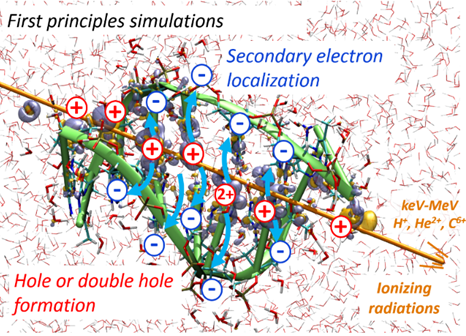
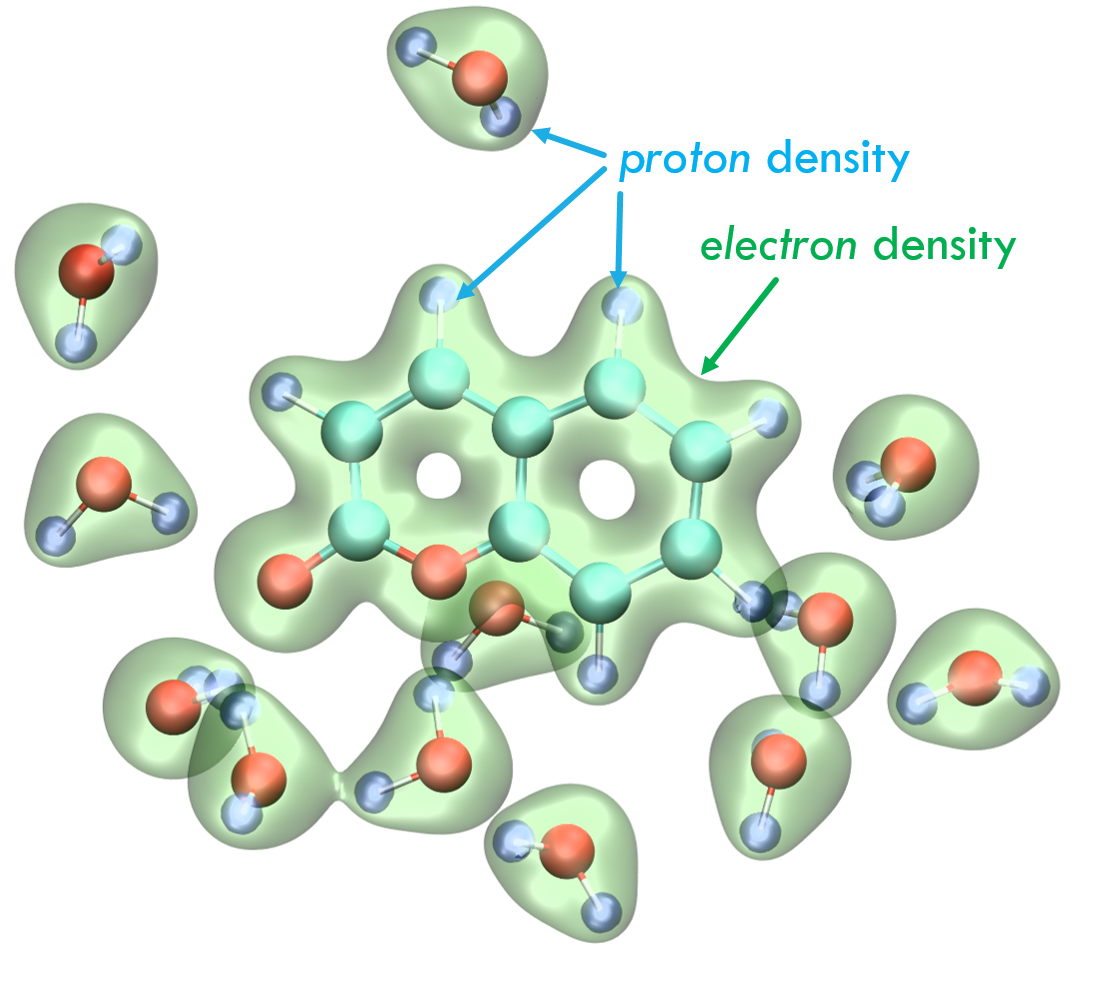
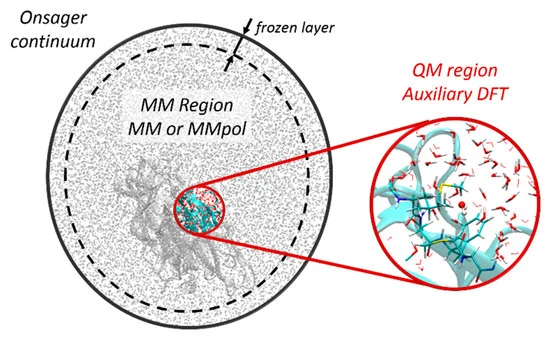
We recently adapted the Auxiliary DFT framework as implemented in deMon2k to the simulation of time-dependent problems via the Runge and Gross equations. Our implementation of the so-called Real-Time-Time-Dependent ADFT (RT-TD-ADFT) fully benefits from the algorithms available in deMon2k to carry out variational density fitting, notably the MINRES algorithm recently proposed for self-consistent-field calculations. We test here MINRES for the first time in the context of RT-TD-ADFT. We report extensive benchmarks calculations to assess the reliability of the ADFT framework. These encompass the construction of absorption spectra in the gas phase and in solvent, the calculation of electronic stopping power curves, the irradiation of zeolites by swift ions and the investigation of charge migrations with attosecond time resolution. All our results are very encouraging. We show that even small auxiliary basis sets are sufficient to obtain results almost undisguisable from those obtained with large and flexible auxiliary bases. Overall, we establish the reliability of RT-TD-ADFT to simulate electronics dynamics in large or very large molecular systems.