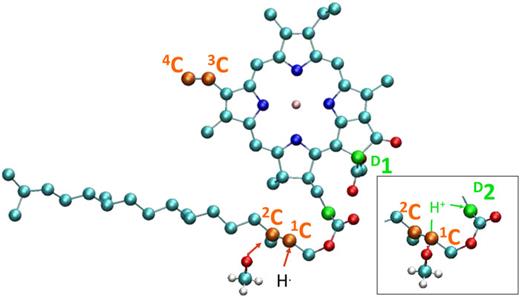
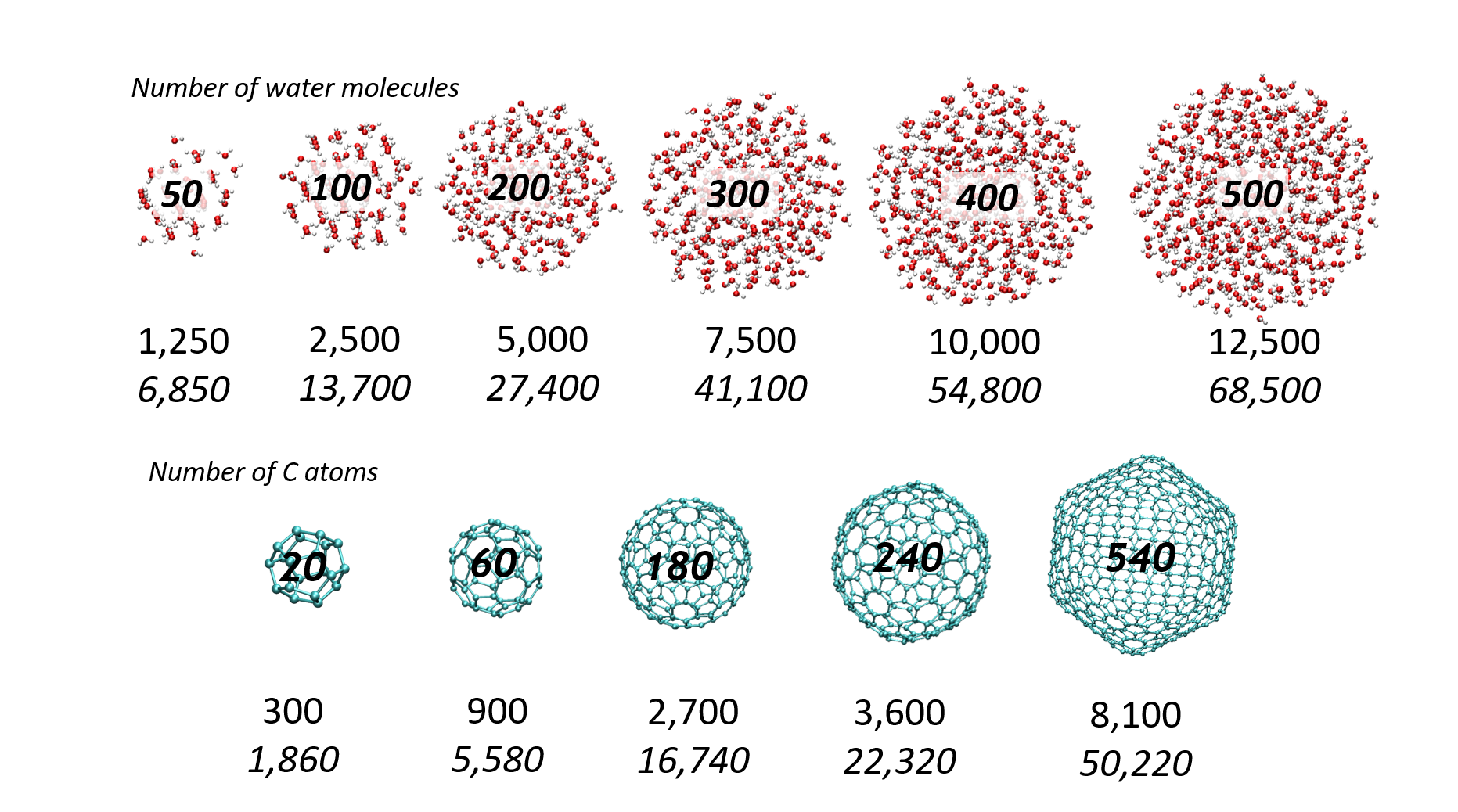
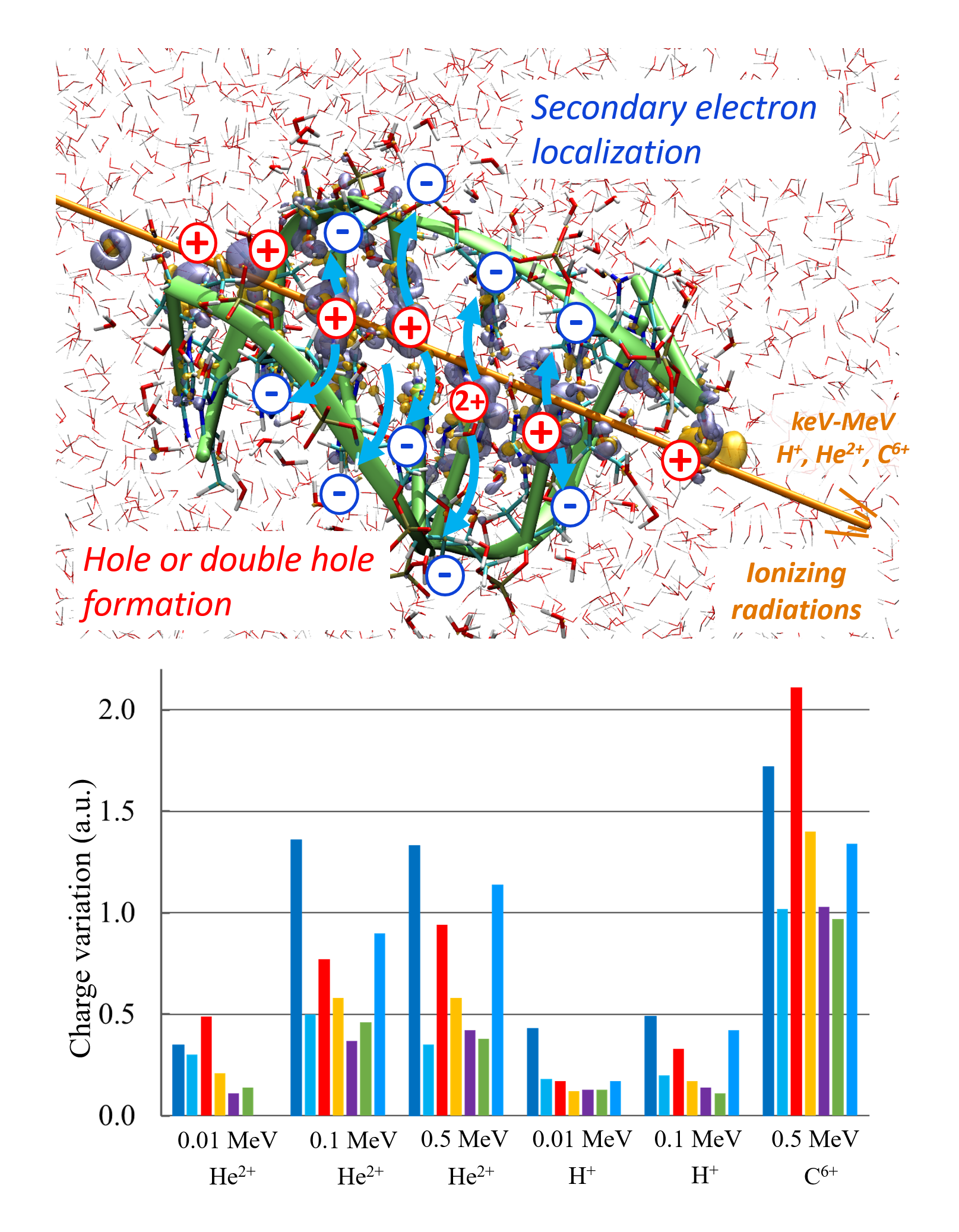
Jean Deviers, Fabien Cailliez, Aurélien de la Lande, Daniel R Kattnig, Comput. Struct. Biol. J. 2024, 26, 11-21. https://doi.org/10.1016/j.csbj.2023.12.009 (open access).
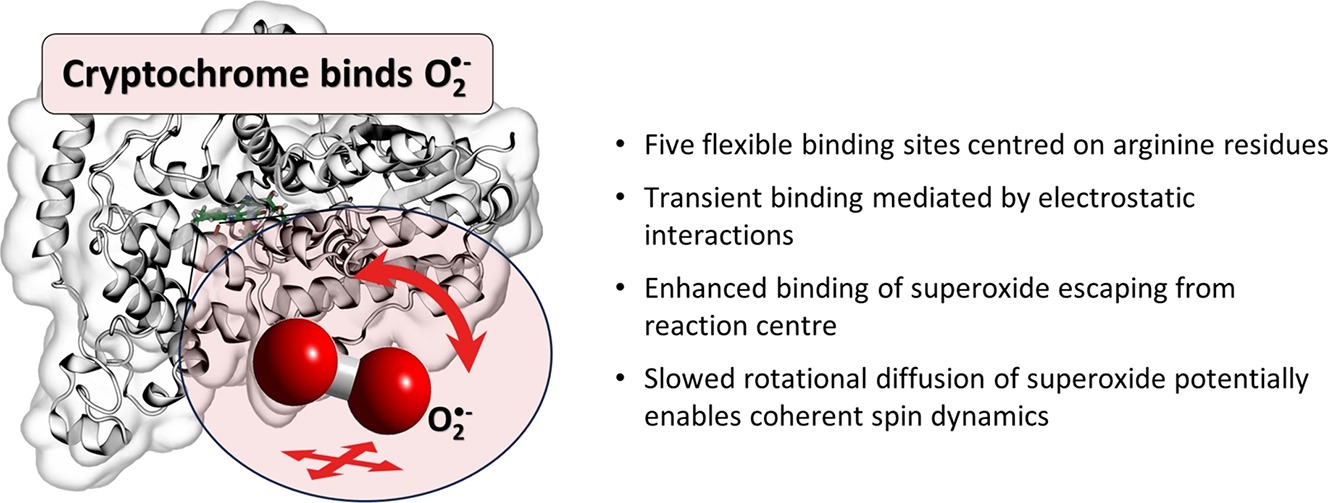
Flavin-binding cryptochromes are blue-light sensitive photoreceptors that have been implicated with magnetoreception in some species. The photocycle involves an intra-protein photo-reduction of the flavin cofactor, generating a magnetosensitive radical pair, and its subsequent re-oxidation. Superoxide (O) is generated in the re-oxidation with molecular oxygen. The resulting O-containing radical pairs have also been hypothesised to underpin various magnetosensitive traits, but due to fast spin relaxation when tumbling in solution would require immobilisation. We here describe our insights in the binding of superoxide to cryptochrome 4 from C. livia based on extensive all-atom molecular dynamics studies and density-functional theory calculations. The positively charged “crypt” region that leads to the flavin binding pocket transiently binds O at 5 flexible binding sites centred on arginine residues. Typical binding times amounted to tens of nanoseconds, but exceptional binding events extended to several hundreds of nanoseconds and slowed the rotational diffusion, thereby realising rotational correlation times as large as 1 ns. The binding sites are particularly efficient in scavenging superoxide escaping from a putative generation site close to the flavin-cofactor, possibly implying a functional relevance. We discuss our findings in view of a potential magnetosensitivity of biological flavin semiquinone/superoxide radical pairs.