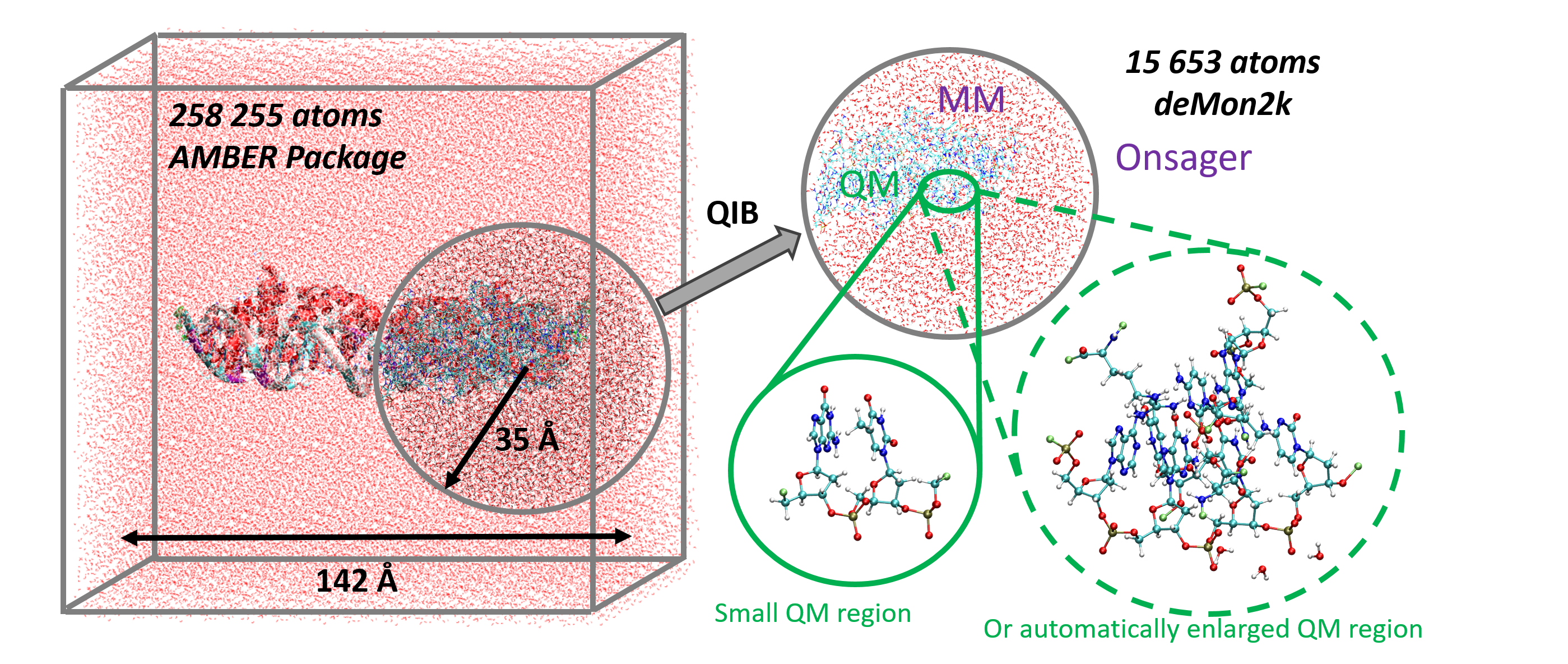
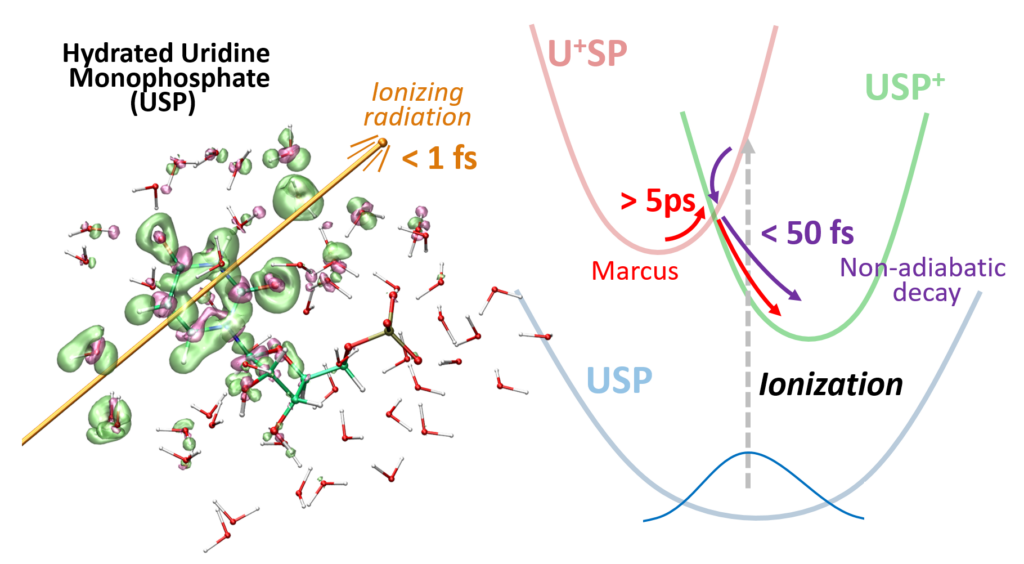
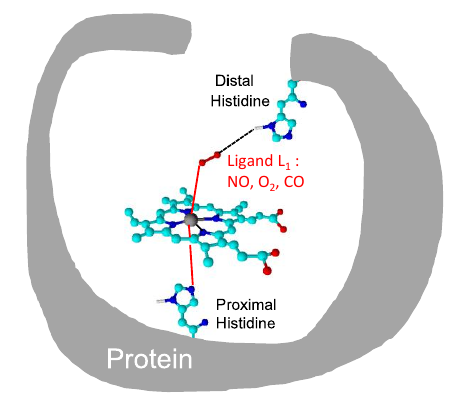
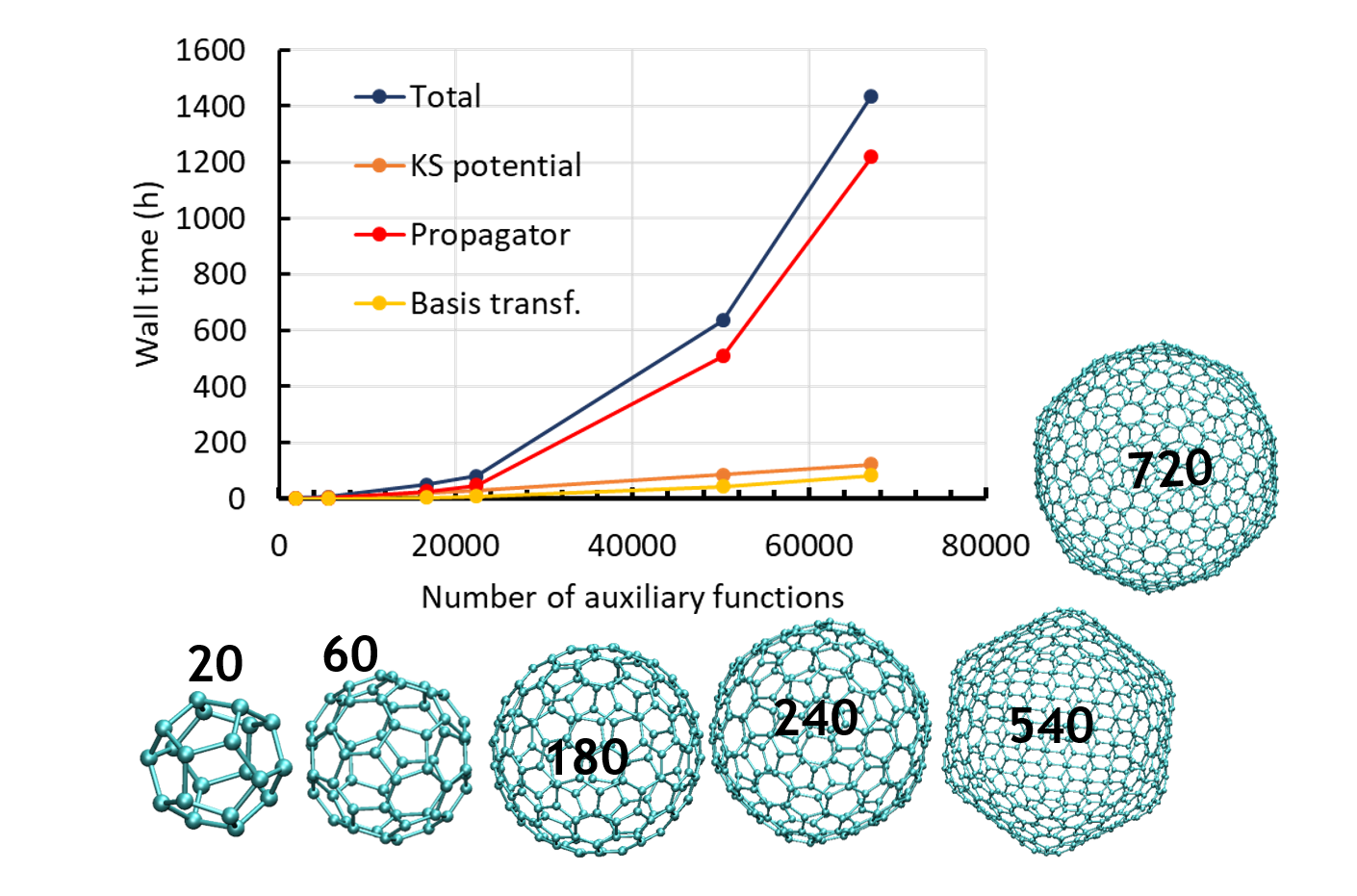
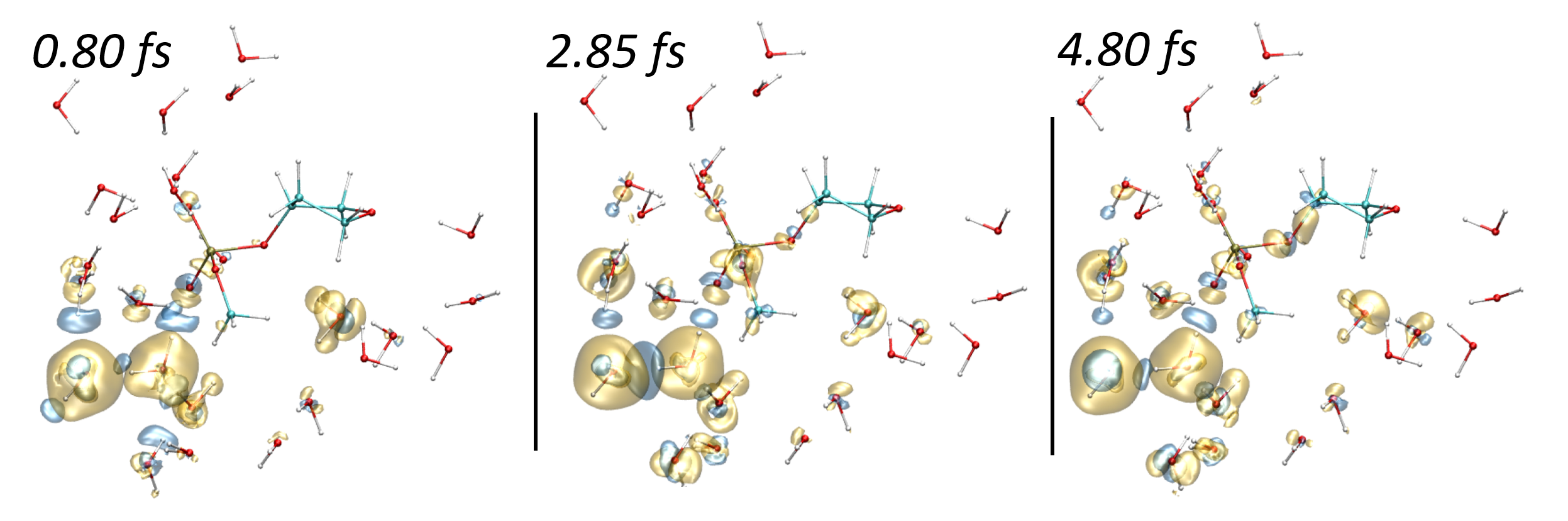
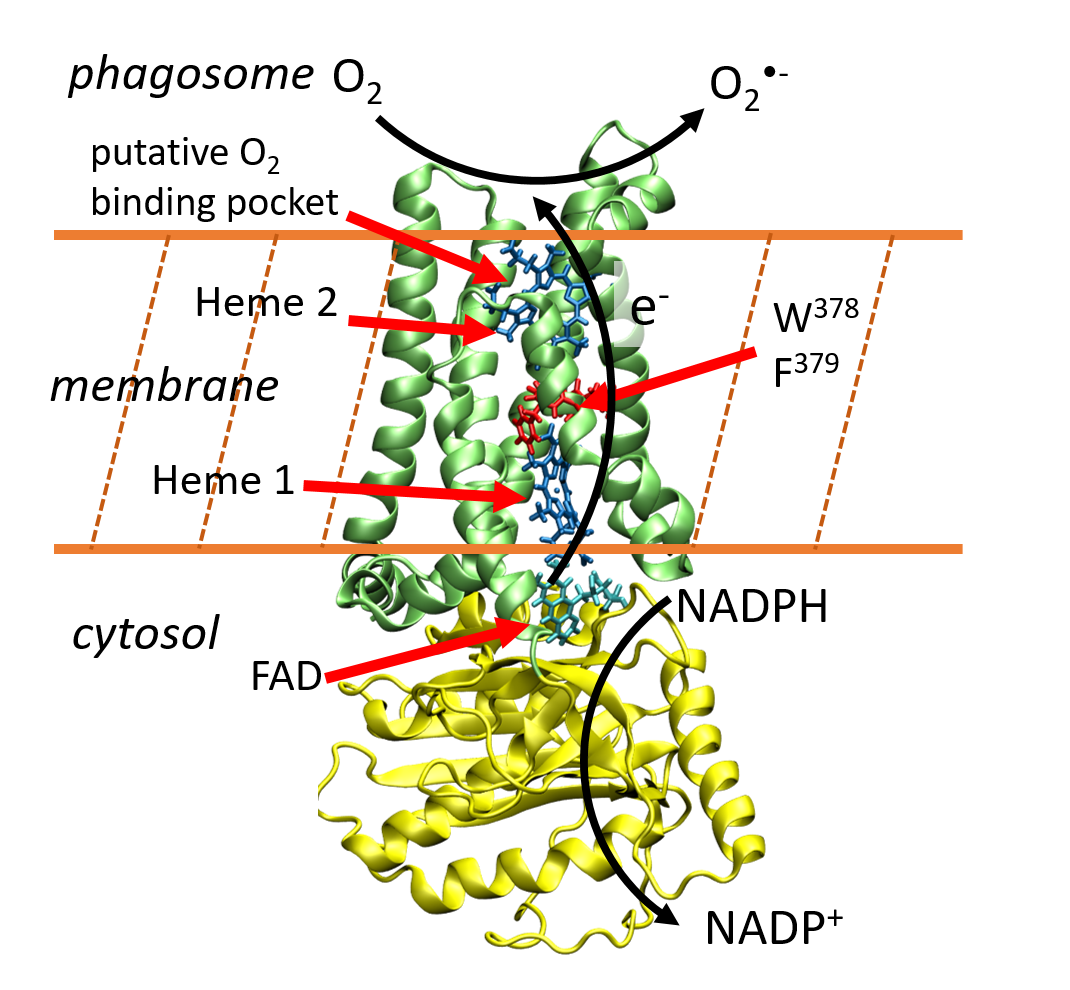
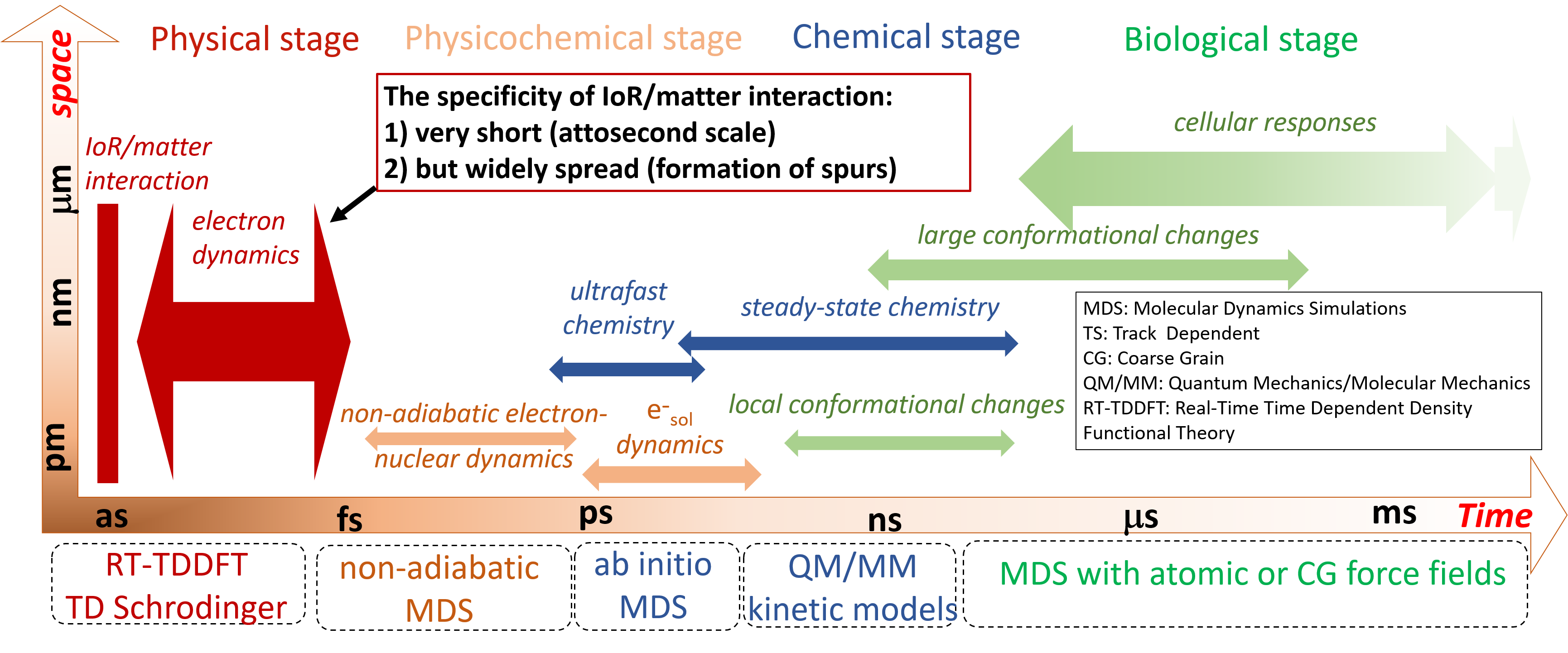
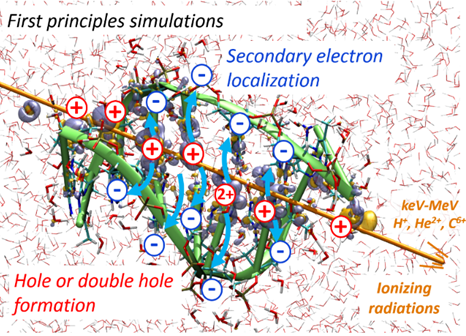
Jean Deviers, Fabien Cailliez, Aurélien de la Lande, Daniel R Kattnig. J. Chem. Phys. 2022, 156, 025101, doi.org/10.1063/5.0078115.
The avian compass and many other of nature’s magnetoreceptive traits are widely ascribed to the protein cryptochrome. There, magnetosensitivity is thought to emerge as the spin dynamics of radicals in the applied magnetic field enters in competition with their recombination. The first and dominant model makes use of a radical pair. However, recent studies have suggested that magnetosensitivity could be markedly enhanced for a radical triad, the primary radical pair of which undergoes a spin-selective recombination reaction with a third radical. Here, we test the practicality of this supposition for the reoxidation reaction of the reduced FAD cofactor in cryptochrome, which has been implicated with light-independent magnetoreception but appears irreconcilable with the classical radical pair mechanism (RPM). Based on the available realistic cryptochrome structures, we predict the magnetosensitivity of radical triad systems comprising the flavin semiquinone, the superoxide, and a tyrosine or ascorbyl scavenger radical. We consider many hyperfine-coupled nuclear spins, the relative orientation and placement of the radicals, their coupling by the electron–electron dipolar interaction, and spin relaxation in the superoxide radical in the limit of instantaneous decoherence, which have not been comprehensively considered before. We demonstrate that these systems can provide superior magnetosensitivity under realistic conditions, with implications for dark-state cryptochrome magnetoreception and other biological magneto- and isotope-sensitive radical recombination reactions.